|
x |
x |
|
 |
 |
|
INFECTIOUS
DISEASE |
BACTERIOLOGIE |
IMMUNOLOGIE |
MYCOLOGY |
PARASITOLOGY |
VIROLOGIE |
|
VIDEO LECTURE |
IMMUNOLOGIE – CHAPITRE DIX-NEUF
IMMUNODEFICIENCES
Abdul Ghaffar, Ph.D.
Emertius Professor of Pathology, Microbiology and Immunology
University of South Carolina
Denis Hudrisier, Ph.D.
Centre national de la recherche scientifique (CNRS) · Institute of
Pharmacology and Structural Biology
Université de Toulouse
|
 |
|
EN ANGLAIS |
Let us know what you think
FEEDBACK |
|
SEARCH |
| |
|
|
 |
|
Logo image © Jeffrey
Nelson, Rush University, Chicago, Illinois and
The MicrobeLibrary |
|
|
|
OBJECTIFS DU COURS
Connaître les immunodéficiences primaires et secondaires
Connaître les immunodéficiences dans le cas du SIDA et d’autres
conditions |
IMMUNODEFICIENCES
L'immunodéficience décrit
l’incapacité du système immunitaire à protéger contre les pathologies
infectieuses ou les cancers.
L’immunodéficience primaire
est provoquée par des anomalies génétiques ou de développement du
système immunitaire. Ces défauts sont présents à la naissance, mais
peuvent se manifester plus tard dans la vie.
L'immunodéficience acquise ou
secondaire est la perte de la fonction immunitaire à la suite d'une
exposition à des agents pathogènes, des facteurs environnementaux, une
immunosuppression ou encore le vieillissement.
|
|
Connaître les
immunodéficiences majeures et leurs caractéristiques
Comprendre les relations entre le site de la lésion et
l’immunodéficience résultante
Connaître les tests de diagnostic pour différentes immunodéficiences
|
IMMUNODEFICIENCES PRIMAIRES
Les immunodéficiences
primaires
sont
provoquées par des défauts génétiques
du système immunitaire
(Figure
1).
Ces défauts peuvent
concerner les mécanismes de défense
immunitaires spécifiques
ou
non
spécifiques.
Ils sont classés
en
fonction de la cible
de la
lésion
lors du
développement ou de
la différenciation
du
système
immunitaire.
Les personnes souffrant
de
déficits immunitaires
sont sensibles à
une variété d'infections
et
le type
d'infection
dépend de la nature
de
l'immunodéficience
(Tableau 1).
|
Table 1. Infections
caractéristiques des immunodéficiences primaires |
|
Composant affecté |
Pathogènes non contrôlés |
Site anatomique affecté |
Exemple clinique |
|
Cellules T |
Bactéries
intracellulaires, virus, protozoaires, champignons |
Non spécifique |
SCID, DiGeorge |
|
Cellules B |
Pneumocoques,
streptocoques, hemophilus
|
Poumons, peau, SNC
|
Déficiences en IgG, IgM |
|
Bactéries entériques et
virus |
Tractus digestif, nez,
yeux |
Déficiences en IgA |
|
Phagocytes |
Staphylocoques,
Klebsielles, Pseudomonas |
Poumons, peau, ganglion
lymphatique drainant |
Maladie granulomateuse
chronique (CGD) |
|
Complément |
Neisseria, Haemophilus,
pneumocoques, streptocoques |
SNC, poumons, peau
|
C3, Facteurs I et H,
composants tardifs du C’ |
|
|
|
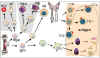 Figure 1
Figure 1
Défauts développementaux dans les immunodéficiences primaires
|
| |
RÉPONSE IMMUNE
SPECIFIQUE
Il existe diverses déficiences
immunitaires qui résultent de défauts dans la différenciation des
cellules souches et peuvent affecter les cellules T, les cellules B et/ou
les immunoglobulines de différentes classes et sous-classes (Tableau 2).
Un défaut dans l'hématopoïèse
précoce affectant les cellules souches conduit à la dysgénésie
réticulaire qui conduit à des défauts immunitaires généraux et une
sensibilité à des infections ultérieures. Cette affection est souvent
mortelle, mais fort heureusement très rare. Elle peut être traitée avec
succès par une transplantation de moelle osseuse.
Immunodéficiences affectant le lignage lymphoïde
Lorsque les cellules progénitrices lymphoïdes sont concernées, alors
ce sont à la fois les lignées de cellules T et B qui sont affectées
et donnent alors lieu à un immunodéficience combinée sévère (SCID,
Severe Combined ImmunoDeficicency). Les nourrissons SCID souffrent
d'infections récurrentes en particulier par les micro-organismes
opportunistes (bactéries, virus, champignons et protozoaires).
Chez environ 50% des
patients SCID, le déficit immunitaire est lié à l'X alors que, pour
l'autre moitié, la déficience est autosomique. Les deux formes se
caractérisent par une absence d’immunité impliquant les cellules T
et B et l'absence complète ou quasi-complète de lymphocytes T et B
circulants. Le thymus est absent à la radiographie.
L’immunodéficience SCID
sévère lié à l'X est due à un défaut dans la chaîne gamma du
récepteur de IL-2, chaîne également commune aux récepteurs de
l'IL-4, -7, -11 et 15, qui sont impliqués dans la prolifération et/ou
la différenciation des lymphocytes. Les SCID autosomiques découlent
principalement de défauts dans les gènes de l'adénosine désaminase
(ADA) ou de la purine nucléoside phosphorylase (PNP), ce qui
entraîne une accumulation respective de dATP ou dGTP, responsables
d’effets toxiques sur les cellules souches lymphoïdes.
D’autres défauts
génétiques conduisant à la pathologie SCID affectent les
recombinases RAG1, RAG2 et la chaîne alpha du récepteur à l’IL-7. En
cas de suspicion de SCID, le patient ne doit pas être vacciné par
des vaccins vivants, au risque de développer une infection par le
pathogène atténué.
Le diagnostic repose sur
la numération des cellules T et B et la quantification des
immunoglobulines. Le déficit immunitaire combiné sévère peut être
traitée par une greffe de moelle osseuse (voir
CMH et
transplantation). Récemment, les patients SCID
autosomiques présentant un déficit en ADA ont été traités avec un
certain succès par une thérapie génique utilisant un vecteur
rétroviral exprimant une version corrigée du gène ADA.
Le phénotype SCID peut
provenir de plusieurs types de déficits
Gènes des recombinases RAG
Les patients ayant une déficience à la fois des cellules T et B
souffrent souvent de déficits en recombinases (RAG1 et 2). Ces
enzymes sont responsables des réarrangements géniques
nécessaires au codage des récepteurs à l’antigène des cellules T
et des immunoglobulines. Ces patients sont athymiques et sont
diagnostiqués sur la base de l’examen des réarrangements de
gènes codant pour le récepteur des cellules T (TCR). Les défauts
liés à l’absence en cellules B ne sont pas observés au début de
la vie du nourrisson en raison de la présence des anticorps
provenant passivement de la mère. Les cellules NK sont normales
chez ces patients. Il s'agit d'un déficit transmissible sur le
mode autosomique récessif.
Chaîne CD3
Chez certains patients SCID, les cellules T peuvent être
présentes mais fonctionnellement défectueuses à cause d’un
défaut de signalisation intracellulaire médié par la chaîne CD3
associée au TCR.
Le récepteur à
l'interleukine-2
La chaîne gamma commune du récepteur à l'interleukine-2
(IL-2Rγc) peut être absente chez certains patients ce qui
empêche de ce fait la signalisation par l’IL-2 ainsi que par
d'autres cytokines agissant comme des facteurs de croissance.
Cela conduit à un défaut dans la prolifération des cellules T,
des cellules B et des cellules NK. Il s'agit d'un déficit
transmissible sur le mode autosomique récessif.
Adénosine
désaminase
L’adénosine désaminase (ADA) est une enzyme responsable de la
conversion de l'adénosine en inosine. Une déficience en ADA
conduit à l'accumulation de l'adénosine ce qui se traduit par la
production de métabolites toxiques qui interfèrent avec la
synthèse d'ADN. Les patients présentent des défauts dans les
cellules T, B et NK.
Les patients SCID
transmettent leur défaut sur le mode autosomique récessif et peuvent
être traités par une thérapie génique ou une transplantation de
cellules souches.
|
Table 2. Résumé des
immunodéficiences affectant les cellules T et B |
|
Pathologie |
Cellules T |
Cellules B
|
Immunoglobulines |
Transmission |
|
Nombre |
Fonction |
IgM |
IgG |
IgA |
|
Dysgénésie réticulaire
|
A |
A |
A |
A |
A |
A |
u |
|
CID (autosomale) |
A/L |
A/L |
A/L |
A/L |
A/L |
A/L |
a |
|
SCID (liée à l’X) |
A/L |
A/L |
A/L |
A/L |
A/L |
A/L |
x |
|
Syndrome de DiGeorge |
A/L |
A/L |
N/V |
N/V |
N/V |
N/V |
a/x |
|
Ataxia telangiectasia |
L |
L |
L |
N/V |
L/V |
L |
a
|
|
Wiskott-Aldrich
|
?V |
L |
L/V |
L |
N |
H |
x |
|
Egalement fort taux d’IgE |
|
Hypogammaglobulinémie liées à l’X |
N |
N |
L |
L |
L |
L |
x |
|
Immunodéficience spécifique en IgA |
N |
N |
N |
N |
L/V |
L |
a/x |
|
Syndrome hypogammaglobulinémique hyper-IgM
|
N |
N |
N |
H |
L |
L |
x |
|
Hypogammaglobulinémie transitoire |
N |
N |
N |
N |
L |
L |
a? |
|
Hypogammaglobulinémie commune variable
(adolescents-adultes) |
N |
N |
N |
N |
L |
L |
Aucune |
|
A: absent; a: autosomique;
H: haut; B: bas; N: normal; I; inconnu; V: variable; x: lié à l’X |
|
|
|
Pathologies des cellules T
Les déficits en cellules T affectent à la fois l’immunité à médiation
cellulaire et l'immunité humorale ce qui rend le patient sensible aux
infections virales, par les protozoaires et champignons. Les infections
virales telles que l’infection par cytomégalovirus et la vaccination contre
la rougeole utilisant un virus vivant atténué peuvent être fatales à ces
patients.
Syndrome de DiGeorge (Délétion sur le
chromosome 22)
Cette immunodéficience des cellules T est la plus clairement définie et
est également connu comme l'aplasie thymique congénitale ou hypoplasie,
ou encore immunodéficience avec hypo-parathyroïdie. Le syndrome est
associé à une hypo-parathyroïdie, une cardiopathie congénitale, une
implantation basse des oreilles et une bouche en forme de poisson. Ces
défauts résultent d’un développement anormal du fœtus (au niveau de la
3e et de la 4e poche pharyngienne) au cours de la 6e à 10e semaine de
gestation lorsque le thymus, les lèvres, les oreilles et l'arc aortique
se forment. Aucune prédisposition génétique n’est claire, et les bébés
atteints du syndrome de Di George ne présentent pas tous une aplasie
thymique. Une greffe de thymus provenant d’un fœtus précoce (13 à 14
semaines de gestation) peut être utilisée pour le traitement. Des
greffes réalisées à partir d’individus plus âgés peuvent provoquer des
réactions de type GVH.
Chez les patients DiGeorge sévèrement immunodéprimés, les vaccins
vivants peuvent causer des infections persistantes.
Le syndrome de Di George se transmet de
manière autosomique dominante (Figure 2) et est causée par une délétion
sur le chromosome 22 (Figure 3). Les délétions sont de taille variable,
mais la taille de la délétion n’est pas corrélée à la sévérité de la
maladie. Dans environ 6% des cas, la microdélétion du chromosome 22 est
héréditaire, mais, dans la plupart des cas, la délétion est provoquée de
novo par des facteurs environnementaux. Les patients peuvent être
traités par une greffe thymique.
|
|
|
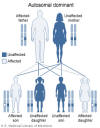 Figure 2
Figure 2
Dans le syndrome de DiGeorge, une délétion sur le chromosome 22q11.2 est
héritée de façon autosomique dominante. National Library of Medicine - NIH
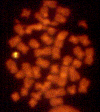 Figure 3
Figure 3
La délétion de gènes dans le syndrome de DiGeorge peut être visualisée par
un signal de fluorescence visible sur une seule des deux copies du
chromosome 22.
David Ian Wilson, University of Newcastle on Tyne - NIH
|
Déficiences en
cellules T accompagnées de déficiences variables en cellules B
L’ataxie-télangiectasie
L’ataxie-télangiectasie est une déficience en cellules T associée à un
défaut de coordination des mouvements (ataxie) et la dilatation des
petits vaisseaux sanguins de la zone de visage (télangiectasie). Les
cellules T et leurs fonctions sont réduites dans des proportions
variables. Le nombre de cellules B et les concentrations en IgM sont de
normale à faible. Les IgG sont souvent réduites et les IgA sont
considérablement réduites (dans 70% des cas). Il existe une forte
incidence de tumeurs malignes, en particulier des leucémies, chez ces
patients. Ces défauts proviennent d’une cassure sur le chromosome 14 au
niveau du locus du TCR et des gènes de chaîne lourde des
immunoglobulines.
Syndrome de Wiskott-Aldrich
Le syndrome de Wiskott-Aldrich est associé à un nombre normal de
cellules T mais présentant des fonctions réduites, qui s'aggravent
progressivement. Les concentrations en IgM sont réduites, mais les taux
d'IgG sont normaux. Les taux d'IgA et d’IgE sont élevés. Les garçons
atteints de ce syndrome développent un eczéma sévère, des pétéchies (due
à un défaut de plaquettes et une thrombopénie). Ils réagissent mal aux
antigènes polysaccharides et sont vulnérables à l'infection purulente.
Le syndrome de Wiskott-Aldrich est une maladie liée à l'X (Figure 4) et
résulte de défauts dans une glycoprotéine du cytosquelette, WASP et de
CD43.
Déficience en CMH (syndrome
des leucocytes « nus »)
Un certain nombre de cas de déficits immunitaires ont été décrits dans
lesquels il existe un défaut dans le gène de la protéine
transactivatrice du CMH de classe II (CIITA), ce qui conduit à un défaut
d’expression en molécules de classe II du CMH sur les APC. Étant donné
que la sélection positive des cellules CD4 dans le thymus dépend de la
présence de ces molécules du CMH, ces patients ont moins de cellules T
CD4 et sont sujets aux infections. Certains patients présentent
également des défauts dans le gène de la protéine TAP et, par conséquent
n'expriment pas les molécules du CMH de classe I conduisant à une
déficience en cellules T CD8+.
|
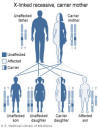 Figure 4
Figure 4
Le syndrome de Wiskott-Aldrich est une immunodéficience liée à l’X
National Library of Medicine - NIH
|
Pathologies des
lymphocytes B
Il existe un certain nombre d’immunodéfieciences dans lesquelles les nombres
et les fonctions des cellules T sont normaux, le nombre de cellules B peut
être faible ou normal, mais les taux d'immunoglobulines sont faibles.
Hypogammaglobulinémie
infantile liée à l'X
L’hypogammaglobulinémie liée à l'X, aussi appelé hypoglobulinémie ou
agammaglobulinémie de Bruton, est l'hypogammaglobulinémie la plus grave
dans laquelle le nombre de cellules B et les taux de toutes les
immunoglobulines sont très faibles. Les patients ont un défaut dans la
maturation des cellules B associé à un défaut du gène codant pour la
tyrosine kinase des cellules B (Btk). Chez ces patients, les cellules B
se développent jusqu’au stade de cellules pré-B possédant les chaînes H
réarrangées d’immunoglobulines, mais pas les chaînes L. Le diagnostic
repose sur une numération des cellules B et la détection des
immunoglobulines. Les patients n'ont pas d’immunoglobulines et souffrent
d'infections bactériennes récurrentes.
Hypogammaglobulinémie
transitoire
A la naissance, les enfants ont des taux d'IgG comparables à ceux de la
mère. Étant donné que la demi-vie des IgG est d'environ 30 jours, le
niveau de ces anticorps d’origine maternelle diminue progressivement,
mais trois mois plus tard les nourrissons commencent à synthétiser leurs
propres IgG. Chez certains nourrissons, cependant, la synthèse d'IgG ne
commence pas avant l'âge de 2 à 3 ans. Ce retard a été attribué à un
défaut d’aide par les cellules T. Il en résulte une insuffisance
passagère en IgG qui peut être traitée par l’administration de γ -globuline
(immunoglobulines).
Hypogammaglobulinémie
variable commune (hypogammaglobulinémie d'apparition tardive)
Ces patients ont des carences en IgG et IgA apparaissant dans la 2ème ou
3ème décennie de leur vie parce que les cellules B ne parviennent pas à
se différencier en cellules plasmatiques. Ces patients sont sensibles à
une variété de bactéries et de protozoaires intestinaux pyogènes. Ils
doivent être traités avec des immunoglobulines administrées par voie
intraveineuse.
Déficience en IgA
Le déficit en IgA est la plus fréquente de toutes les immunodéficiences
(1/700 de tous les individus caucasiens) et fait suite à un défaut de
commutation de classe. Environ 20% des personnes atteintes de déficit en
IgA ont également de faibles taux en IgG. Les patients déficients en IgA
sont très sensibles aux infections gastro-intestinales, oculaires et
rhino-pharyngées. Les patients atteints de déficit en IgA ont une
incidence élevée de maladies auto-immunes (notamment celles mettant en
jeu des dépôts de complexes immuns) et de tumeurs malignes lymphoïdes.
Des anticorps anti-IgA (IgG) sont détectés dans 30 à 40 pour cent des
patients qui ne devraient pas être traités avec des γ-globulines. Le
diagnostic de laboratoire est basé sur la mesure IgA.
Déficit sélectif en IgG
Des carences en différentes sous-classes d'IgG ont été trouvées chez des
patients. Ces patients sont sensibles aux infections pyogènes.
Immunodéficience hyper-IgM
liée à l'X
Les personnes atteintes de ce type d’immunodéficience ont de faibles
concentrations en IgA et IgG avec des niveaux anormalement élevés d'IgM.
Ces patients ne peuvent pas faire de commutation de classe d'IgM vers
d'autres classes, ce qui est attribué à un défaut d’expression de CD40L
sur leurs cellules T CD4+. Ils sont très sensibles à l'infection
purulente et doivent être traités avec des γ-globulines intraveineuses.
|
 Figure 5
Figure 5
Faible élimination intracellulaire des bactéries dans la maladie
granulomateuse chronique |
SYSTEME IMMUNITAIRE
NON-SPÉCIFIQUE : DÉFICIT AFFECTANT LE LIGNAGE MYELOIDE
Il existe des
immunodéficiences primaires du système immunitaire non spécifique. Elles
consistent essentiellement en défauts portant sur les cellules
phagocytaires, les cellules NK et le système du complément.
Agranulomatose
congénitale
Dans ce cas, les patients ont une diminution du nombre de
neutrophiles. Cela est dû à un défaut de différenciation des
cellules progénitrices myéloïdes en neutrophiles. Ces patients sont
traités avec une cytokine : le GM-CSF (Granulocyte-Macrophage Colony
Stimulating Factor) ou le G-CSF.
Défauts du système
phagocytaire
Des défauts de cellules phagocytaires (nombre et/ou fonctions)
peuvent conduire à une susceptibilité accrue à une variété
d'infections.
Neutropénie cyclique
Cette immunodéficience se traduit par un faible nombre de
neutrophiles circulants retrouvé environ toutes les trois semaines.
La neutropénie dure environ une semaine au cours de laquelle les
patients sont sensibles aux infections. Le défaut semble être du à
une mauvaise régulation de la production des neutrophiles.
Maladie granulomatose
chronique (CGD)
La CGD se caractérise par une adénopathie marquée, une
hépato-splénomégalie et une inflammation chronique des ganglions
lymphatiques drainants. Les leucocytes ont des mécanismes
microbicides intracellulaires peu efficaces (Figure 5) et présentent
une faible production d’espèces réactives de l’oxygène. Chez la
majorité de ces patients, le déficit est dû à un défaut de la NADPH
oxydase (cytochrome b558: gp91phox, ou plus rarement de la gp22phox)
ou d'autres protéines agissant comme cofacteurs (gp47phox, gp67phox)
participant au métabolisme oxydatif des cellules phagocytaire. Ces
patients peuvent être diagnostiqués sur la base d'une mauvaise
réaction de réduction du nitro-bleu de tétrazolium (NBT) qui est une
mesure de la stimulation du métabolisme oxydatif. Le traitement de
ces patients par l'interféron-gamma a été couronné de succès.
|
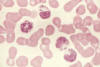 Figure 6
Figure 6
La photo est tirée d’un patient présentant le syndrome de Chediak-Higashi.
D’énormes granules sont détectés dans le cytoplasme des granulocytes. Ils
proviennent de la fusion anormale de granules au cours de leur formation.
Ces granules anormaux sont retrouvés dans de multiples types cellulaires
dans tous le corps.
National Cancer Inst
|
Déficience dans l’adhérence
leucocytaire
Dans cette maladie, les cellules T et les macrophages n’expriment pas le
récepteur du complément CR3 en raison d’un défaut d’expression de l’un
ou l’autre des polypeptides CD11 ou CD18 composant ce récepteur et, par
conséquent, ils ne peuvent pas réagir avec l’opsonine C3b. Il peut
également y avoir un défaut dans les molécules d’adhérence LFA-1 ou
Mac-1 de la famille des intégrines, résultat de l’expression déficiente
des polypeptides CD11a ou CD11b entrant dans la composition de ces
récepteurs respectivement. Ces molécules sont impliquées dans la
diapédèse et donc, les neutrophiles déficients ne peuvent pas répondre
efficacement aux signaux chimiotactiques. Le traitement consiste en une
transplantation de moelle osseuse (déplétée en cellules T et compatible
au niveau du CMH) ou la thérapie génique.
Syndrome de Chediak-Higashi
Le syndrome de Chediak-Higashi est marqué par la réduction (cinétique
ralentie) de la destruction intracellulaire des pathogènes et du
chimiotactisme accompagnée par un défaut de la fusion des phagosomes
avec les lysosomes ainsi qu’une carence en protéases. Des lysosomes
(granules intracellulaires) géants sont souvent identifiés dans ces
cellules (Figure 6). La production de radicaux oxygénés est normale.
Cette immunodéficience est souvent accompagnée de défauts dans les
cellules NK et les plaquettes et des troubles neurologiques sont notés.
|
| |
PATHOLOGIES DU SYSTEME
DU COMPLEMENT
Des anomalies du système du
complément conduisent également à une susceptibilité accrue aux infections.
Il existe des déficiences génétiques de différents composants du système du
complément, dont le plus grave est la carence en C3 qui pourrait résulter
d’une synthèse faible de C3 ou d'une déficience en facteur I ou en facteur
H.
|
| |
IMMUNODEFICIENCES
SECONDAIRES (ACQUISES)
Immunodéficiences
associées à des infections
Les infections bactériennes, virales, par les protozoaires, les
helminthes et les champignons peuvent conduire à des carences en
cellules B, en cellules T, en macrophages et/ou en PMN. Parmi ces
pathologies, le syndrome d'immunodéficience acquise (SIDA) est le plus
marquant. Des immunodéficiences secondaires sont également observées
chez les patients porteurs de tumeurs malignes.
Défauts
immunologiques dans le SIDA
Les diverses immunodéficiences acquises passent inaperçue en regard du
sida qui est causée par le virus de l’immunodéficience humaine (VIH-1).
Ce virus a été découvert en 1981 et les patients présentaient des
infections fongiques opportunistes avec des organismes tels que
Pneumocystis carinii et dans d'autres cas, une tumeur de la peau connue
sous le nom de sarcome de Kaposi. Il existe deux grands types de VIH: le
VIH-1 et 2, le premier étant la souche trouvée fréquemment en Amérique
du Nord. Le VIH se transmet par les rapports sexuels, le sang infecté et
les fluides corporels ainsi que de la mère au fœtus. Le VIH est un
rétrovirus à ARN lequel subit une transcription inverse en ADN utilisant
la transcriptase inverse virale (RT) après que l'entrée du virus dans la
cellule ait eu lieu. L'ADN est intégré dans le génome de la cellule hôte
en tant que provirus et est répliqué en même temps que le génome de la
cellule hôte. Le VIH-1 ne se réplique pas dans la plupart des autres
animaux, mais infecte les chimpanzés chez qui il n'induit pas le sida.
Les souris SCID reconstituées avec des lymphocytes humains peuvent aussi
être infectés par le VIH-1. Le virion du VIH-1 se compose d'une
enveloppe virale constituée d’une bicouche lipidique externe issue de la
cellule hôte dans laquelle sont intégrées les glycoprotéines virales
constituées de la gp41 transmembranaire associées à la gp120. La gp120
se lie au récepteur CD4 exprimé sur les cellules hôtes. Protégée par
l'enveloppe virale on retrouve la nucléocapside constituée d'une couche
de protéine de matrice composée de p17 et une capside interne constitué
de p24. Le génome viral est constitué de deux molécules d'ARN simple
brin associées à deux molécules de RT ainsi qu’à d'autres enzymes,
incluant une protéase et une intégrase.
Cycle de réplication et
cibles de la thérapie
Le virus se fixe à la molécule CD4 sur les cellules Th, les
monocytes et les cellules dendritiques par l'intermédiaire de la
gp120 du VIH. Pour l'infection par le VIH, un corécepteur est
nécessaire. Le corécepteur est un récepteur de chimiokine comme
CXCR4 ou CCR5. CCR5, principalement exprimé sur les macrophages, et
CXCR4, sur cellules T CD4+, servent de corécepteurs pour l'infection
à VIH. Après la fusion de l'enveloppe du VIH et de la membrane de
l'hôte, la nucléocapside pénètre dans la cellule. La RT synthétise
l'ADN viral, qui est transporté vers le noyau où il s'intègre dans
l'ADN de la cellule sous la forme d'un provirus. Le provirus peut
rester latent jusqu'à ce que la cellule soit activée et que le
provirus subisse alors également la transcription. Les virions,
comprenant de l'ARN et des protéines virales transcrites sont alors
produits. Ces particules bourgeonnent à la membrane de la cellule
hôte où ils acquièrent l'enveloppe créant ainsi de nouveaux virus
infectieux. Sur la base de ces connaissances, des agents
thérapeutiques ont été mis au point qui ciblent l'entrée du virus
(fusion des membranes du virus et de l’hôte) ainsi que des
inhibiteurs d’enzymes telles que la RT, la protéase et l’intégrase
virales. La thérapie antivirale hautement active (highly active
anti-retroviral therapy, HAART) est un cocktail de trois ou plus de
ces agents.
Perturbations
immunologiques
Le virus se réplique rapidement et, en environ deux semaines, le
patient peut développer une fièvre. La charge virale dans le sang
augmente de manière significative et atteint sont pic en deux mois,
après quoi il y a une baisse soudaine liée à la présence du virus
latent retrouvé dans les centres germinatifs des ganglions
lymphatiques. Les CTL antiviraux se développent très tôt alors que
les anticorps peuvent être détectés entre 3 et 8 semaines.
L’élimination par les CTL des cellules Th infectées se produit
autour de 4 - 8 semaines après l’infection et conduit à une
diminution progressive des cellules T CD4+. Lorsque le nombre de
lymphocytes T CD4 + diminue en dessous du seuil de 200 par
millimètre cube, un sida se développe.
Immunothérapie
Il y a plusieurs limites au développement d'un vaccin efficace
contre le VIH.
-
Le vaccin atténué peut
induire la maladie
-
Les cellules T CD4+
peuvent être détruites par le vaccin
-
La variabilité
antigénique du VIH
-
Une faible
immunogénicité du virus lié à la baisse d’expression des
molécules du CMH
-
Le manque de modèles
animaux
-
Le manque de tests
in vitro
Les approches suivantes
ont été adoptées pour les vaccins en cours de développement
-
Vaccination avec des
mutants de délétion visant à réduire la pathogénicité
-
Vaccination avec des
protéines recombinantes
-
Vaccination avec des
vecteurs viraux exprimant des gènes codant pour des protéines du
VIH
-
Administration de
chimiokines agissant comme des compétiteurs pour la liaison du
VIH-1 aux corécepteurs
-
Administration d’IL-2
dans le but de stimuler les cellules Th.
Immunodéficiences liées au
vieillissement
Ces immunodéficiences comprennent une involution progressive du cortex
thymique avec une baisse de la cellularité et une réduction de la taille
du thymus, une diminution de la fonction des cellules suppressives et
par conséquent une augmentation de l'auto-réactivité et, enfin, une
diminution des fonctions des cellules T CD4+. A l’inverse, les fonctions
des cellules B peuvent être légèrement accrues.
Immunodéficiences
associées au cancer et autres pathologies
Des déficits en cellules B ont été notés dans le myélome multiple, la
macroglobulinémie de Waldenstrom, la leucémie lymphoïde chronique et les
lymphomes bien différenciés. La maladie de Hodgkin et les tumeurs
solides au stade avancé sont associées à une insuffisance fonctionnelle
des cellules T. La plupart des agents chimiothérapeutiques utilisés pour
le traitement des tumeurs malignes sont également immunosuppresseurs.
D'autres conditions dans
lesquelles se produisent des immunodéficiences secondaires sont l'anémie
falciforme, le diabète sucré, la malnutrition protéino-calorique, les
brûlures, la cirrhose alcoolique, la polyarthrite rhumatoïde, un
dysfonctionnement rénal, etc…
|
|
|
 Retourner à la section d'immunologie de Microbiologie et Immunologie On-line
Retourner à la section d'immunologie de Microbiologie et Immunologie On-line
This page last changed on
Monday, October 06, 2014
Page maintained by
Richard Hunt
Please report any problems to
richard.hunt@uscmed.sc.edu
|
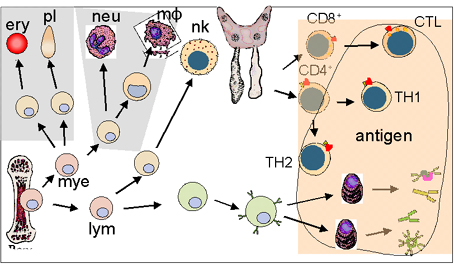 Figure 1
Figure 1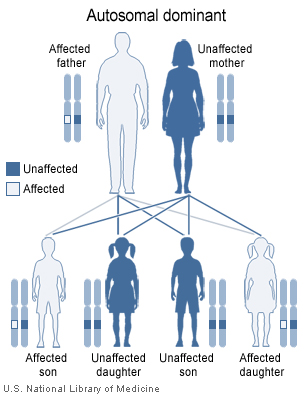 Figure 2
Figure 2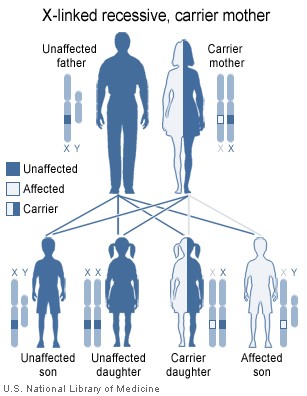 Figure 4
Figure 4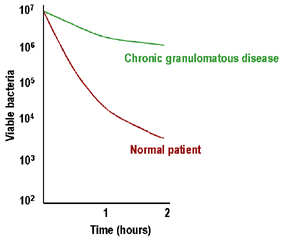 Figure 5
Figure 5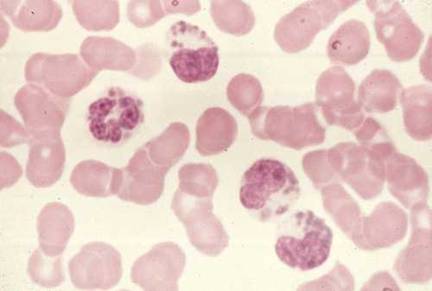 Figure 6
Figure 6