![]()

VIROLOJİ - BÖLÜM YİRMİÜÇ
SİNİR SİSTEMİNİN YAVAŞ VIRUS HASTALIKLARIDr Margaret Hunt
Professor Emerita
Department of Pathology, Microbiology and Immunology
University of South Carolina School of Medicine
Columbia
Çeviren
Prof. Dr. Selçuk Kaya
İzmir Katip Çelebi Üniversitesi, Tıp Fakültesi
FEEDBACK
Dr Richard Hunt tarafından gösterildi ve düzenlendi
ÖĞRENME HEDEFLERİ
Merkezi sinir sisteminin subakut/kronik hastalıklarına giriş yapmak.
Merkezi sinir sisteminin subakut hastalıkları ile ilişkili geleneksel
olmayan ajanların özellikleri
GENEL
"Yavaş virüs enfeksiyonları" ifadesi hastalığın gidişatına
işaret etmektedir, virüsün gelişim hızına değil. Bu hastalıklar uzun bir
inkübasyon periyoduna (ki bu aylar ya da yıllar olabilir) ve uzun süreli,
progresif klinik gidişe sahip olabilir.
Yavaş virüs enfeksiyonları sık görülen ya da sık görülmeyen virüs (özellikle sık
görülmeyen ajanlar veya atipik virüs/ajanlar) kaynaklı olabilir.
Santral sinir sisteminin yavaş virüs/prion hastalıkları ilişkili
semptomları çoklu nörolojik görünüme eğilimli olurlar. Farklı hastalarda çok
farklı semptomlarla görülebilirler.
Şekil 1
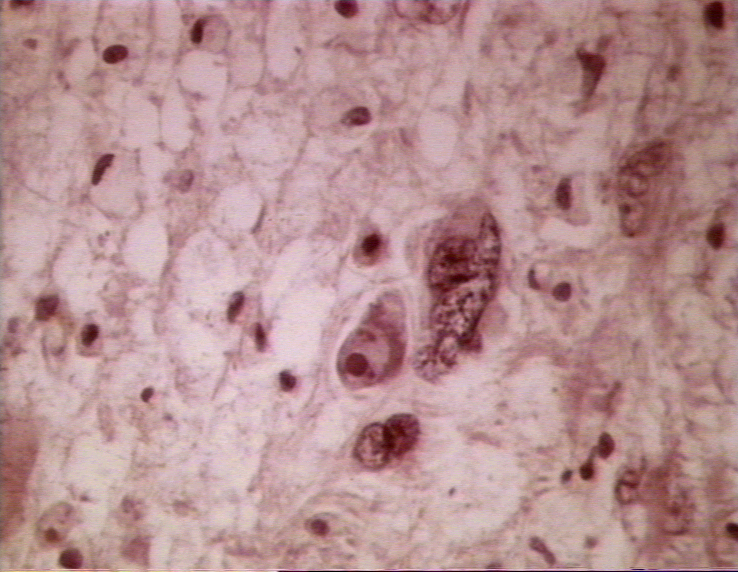
Beyinde progresif multifokal lökoensefalopati
 Şekil 2
Şekil 2
Beyinde progresif multifokal lökoensefalopati
© Bristol Biomedical Image Archive. Used with permission
Progresif multifokal lökoensefalopati (PML)
SSS nin oligodendrositleri öldüren, nadir, ilerleyici, fatal ve demiyelinizan bir hastalığıdır (Şekil 1,2). Hafıza kaybı, koordinasyon kaybı, mental problemler, görme problemleri ile sonuçlanır.
Bu hastalık poliomavirüs ailesinin belirli üyeleri, genellikle JC virüsten kaynaklanır. Seroloji JC virüs maruziyetinin yaygın olduğunu göstermekte ancak PML nadirdir. PML gelişen hastalar sıklıkla immün sistem anormalliklerine sahiptir. AIDS li hastaların %5 inde PML gelişir. HAART bu hastaların az bir kısmını ve bu hastalarda nöroradyolojik tablo gelişimini stabilize edebilir. Her nasılsa HIV pozitif PML hastalarının hepsinde olmamak üzere HAART tedavisi alanlarda PML de belirgin bir yanıt görülür. PML latent JC virüs enfeksiyonun (muhtemelen böbrekteki) reaktivasyonundan ileri gelebilir. Beyinde bile çokça virüs vardır.
Diğer bir polyomavirüs olan BK virüs böbreklerde latent enfeksiyon, immün sistem baskılanması altında reaktivasyon ve çeşitli üriner sistem enfeksiyonları oluşturabilr. PML ile ilişkisi yoktur. 2006 da BK virüsün prostat kanserinde rol oynayabileceği ileri sürülmüştür.
Subakut sklerozing panensefalit (SSPE)
Kızamık hastalığının nadir bir komplikasyonudur ve primer enfeksiyondan yaklaşık olarak 1 ila 10 yıl sonra gelişir. İlerleyici ve fataldir ve motor-mental bozulma ile karakterizedir. Risk faktörleri primer kızamık hastalığının erken yaşta kazanılması ile artar.
SSPE beyindeki defektif virüs formları ile ilişkilidir ve virüs ile infekte hastalardan izolasyon zordur. Kızamık aşılanmasından beri insidansı azalmaktadır.
Progresif rubella panensefalit (PRP)
PRP rubella virüs enfeksiyonlarının çok nadir bir sonucudur ve motor -mental bozulma ile sonuçlanır. Enfeksiyon başlangıcı genellikle konjenital veya doğum sonrası ve PRP başlangıcı 8-19 yaşarası meydana gelir. Hastalık kliniği birkaç yılın üzerinde uzayabilir.
Diğer yavaş virus infeksiyonları
Human immunodeficiency virus ve AIDS. Bakınız HIV/AIDS bölümü
Rabies. Bakınız rabies bölümü
PRIONLAR
 Şekil 3
Şekil 3Geleneksel virus ve prionların özelliklerinin karşılaştırılması
SSS nin yavaş hastalıklarından bazıları sık görülmeyen varlığı hala tartışmalı bir grup ajan tarafından meydana getirilir. Bir şekilde konvansiyonal virüslere benzerler: Oldukça küçük, filtre edilebilir, büyümek için konağın hücrelerine ihtiyaç duyan ajanlardır. Enerji üretimi ve protein sentez kapasiteleri yoktur.
Diğer yönden her nasılsa virüslerden oldukça farklılar.
Örneğin: infekte doku veya infekte materyalden hazırlanan preperatlarda
virüs partiküllerinin hiçbir belirtisini göremeyiz. Bu ajanların nükleik
asit içerdiğini kimse kanıtlayabilmiş değil. Eğer nükleik asit içeriyorlarsa
çok küçük ve çok az kopyalama kapasitesine sahip. Bu ajanlar virüsleri
inaktive etmek için yaygınca kullanılan tedavilere beklenmedik bir dirence
sahipler (Şekil 3,4).
 Şekil 4
Şekil 4
Geleneksel virusların özellikleri
 Şekil 5
Şekil 5
Bulaşıcı spongioform ensefalopati
 Şekil 6
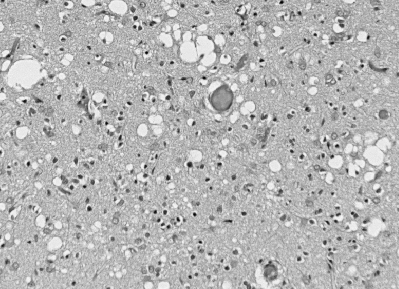
Şekil 6
CJD'de spongiform değişiklik, kortikal hücre mimarisini bozan, hem tek
başına hem de birleşik gruplarını oluşturan neuropil içinde çok sayıda
yuvarlak vakuollerle oluşur.
© The UK Creutzfeldt-Jakob Disease Surveillance Unit
İnfekte materyalden alınan örneklerde protein varlığından ve
proteini yok eden tedavilerin infeksiyonu da yok etmesinden dolayı bu
konvansiyonal olmayan virüs veya ajanlara ‘prion’ adı verilir. Buna karşın
nükleik asitleri yok eden tedaviler infeksiyonu yok etmezler. Söz konusu
protein, Prion protein(PrP) olarak bilinmekte. Bu ajanların bir kısmında nükleik
asit olup olmadığı hala tartışmalı bir soru. Çoğu insan hala infekte materyelin
yalnızca bir protein olduğunu düşünmekte.
Bu ajanların:
-
oluşturduğu hastalıklar SSS'ne sınırlıdır
-
uzun inkübasyon süresine sahiptir
-
yavaş, progresif ve fatal bir hastalık seyri gösterir
-
spongioform bir ensefalopati izlenir
-
karakteristik olarak nöronlarda vakuolizasyonla sonuçlanır
-
amiloid benzeri karaktere sahip ve PrP içeren fibriler agregat formları oluşumu meydana getirebilir
Bu ajanlar tarafından meydana gelen hastalıklar(taşınabilir/bulaşıcı
spongioform ensefalitler (Şekil 5)) insanlarda oldukça nadirdir ancak
düşünüldüğünden daha yaygın olabileceğine dair spekülasyonlar ve SSS nin diğer
dejeneratif hastalıkları ile ilgili çalışma sonuçları vardır. Edinilmiş,
kalıtımsal veya sporadik meydana gelmiş olabilir
Hayvanlarda Prion-sebepli Hastalıklar
Scrapie
Scrapie koyunların bir hastalığıdır. Davranış değişiklikleri, progrese tremor, ataksiler (kas koordinasyon kaybı), wasting ve ölümle sonuçlanır. BulaşıcI bir hastalıktır.
Sığır Spongiform Ensefalit (BSE)
BSE; diğer bir deyişle deli dana hastalığı olarak bilinen bu hastalık, büyük baş hayvanlarda progresif nörolojik dejenerasyonla sonuçlanan prion kaynaklı bir hastalıktır. İlk vaka 1986 da birleşik krallıkta belirlenmiş, infeksiyon belkide 1970’li yıllarda ortaya çıkmıştı. Bulaşma muhtemelen, BSE ile enfekte diğer sığırların ( enfeksiyon spontan meydana gelmiş olabilir) sığırların et ve kemikle beslenmesi veya Scrapie ile infekte koyunların et ve kemikleriyle beslenmesi sonucu meydana gelmekte. Bu uygulamaların yasaklanması ile hastalık kontrolü sağlanmıştır.Birleşik krallık ta 1992 de haftada binden fazla yeni vaka raporlandığında, BSE pik yapmıştı ancak hastalığın kontrolü sonucu yeni infeksiyon hızı yılda 1-2 ye kadar düşmüştür.
Kuzey amerika da Şubat 2015 den beri 24 vaka olmuş, bunların 20 si Kanada da.
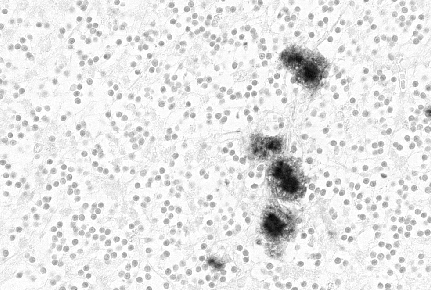 Şekil 7
Şekil 7Amiloid plaklar gösteren prion protein immunboyama
© The UK Creutzfeldt-Jakob Disease Surveillance Unit
İnsanlarda Prion-sebepli Hastalıklar
Kuru
Kuru insanların bir hastalığı. Tremor ve ataksi ve sıklıkla ilerleyen evrelerde demansa yol açar. Papua/Yeni Gine’deki Fore halkında yamyamlık ve ölülerin otopsisini içeren din törenleriyle bulaşır. Bu uygulamalar ortadan kalktığından beri doğmuş hiç kimse Kuru geçirdi mi? Fetusa geçtiğine, süt veya yakın temas ile bulaşına dair bir kanıt yok.
Creutzfeldt-Jakob disease (CJD)
CJD insanların, demans, sıklıkla tremor ve motor koordinasyon kaybı ile sonuçlanan bir hastalığı. Amerika Birleşik Devletleri'nde her milyon popülasyonda yıllık 1 -2 vakaları var ancak tanı konulmamış vakalar olabilir. Bu hastalık laboratuarda hayvanlara bulaşmış olabilir. Hastalığın 16-80 yaşları arasında gelişebildiği gözlemlenmiş olsa da sıklıkla 50-70 yaşlarda görülür. Vakaların %10 u, büyük olasılıkla açıkca CJD gelişimi yapmakta olan bireylerde bir genin görüldüğü familiyal kökenlilerdir.
Sık bulaş yolları bilinmemekte ancak çoğu vaka sporadik ve kişiden kişiye direk bulaşla ilgili kanıt yok. CJD, kornea nakli, dura mater transplantı, nöroşirurjide uygunsuz şekilde sterilize edilmiş ekipmanın kullanımı (sterilizasyon prosedürleri şimdi bunu önlemek için değiştirildi), kadavra kaynaklı insan growth hormon uygulanması (şuan insan GH yapımında Rekombinan DNA vektörleri kullanılmakta) gibi medikal manipülasyonlar aracılığı ile bulaşmış olabilir.
Yeni varyant CJD hastalığı (insan BSE); nvCJD, vCJD
CJD nin yeni bir formu 1996 da öncelikle Birleşik Krallıkta raporlanmış. Hastalar çoğu CJD hastasından (ortalama ölüm yaşı 68) daha genç imiş (sıklıkla 40 ın altında; ortalama ölüm yaşı 28 yaş)(Şekil 8A) Bu hastalık psikiyatrik problem varlığı ve hastalık seyrinin daha uzun süreli olma eğilimden dolayı da genel CJD ından farklıdır. Bu hastalık varyant Creutzfeldt-Jakob hastalığı veya vCJD olarak bilinir.
Varyant CJD hastaları zamanla diğer insan prion hastalıklarında tanımlanmış tüm semptomları gösterebilir veya hiçbir semptom göstermeyedebilir. BSE kontamine et maruziyeti ile ilişkili olduğuna dair güçlü kanıtlar vardır. Şuan güçlü BSE kontrol önlemleri uygulanmaktadır. Otopsiler ayırıcı nöropatolojik bir görünüm ve tipik CJD vakalarından daha fazla PrP amiloid plak tipli depozitler göstermektedir (Şekil 7,9).
Klasik ve variant CJD arasındaki farklar BSE vakalarının sayısı
2014 te Tüm dünyada 220 yi aşkın raporlanmış varyant CJD vakası bulunmakta idi. UK’de 177 insan v CJD li olarak raporlanmış olup bunların hepsi ölmüştür (Şekil 8b). 27 Fransa'da, 4 İrlanda'da, 4 Amerika Birleşik Devletleri' nde, Kanada, Japonya, Portekiz, İspanya ve Hollanda'da herbirinde 1 (Kanada, Japonya ve US den olan insanlar muhtemel UK de yaşarken maruz kalmışlar) UK de epideminin pik yaptığı yıl ; 27 insanın vCJD li olarak tanı aldığı ve bu hastalıktan 28 ölümün olduğu 2000 yılı idi.
Büyük bir salgın başlangıcı görürsek hastalıklı bu vakaların çoğunluğuna ne olacağını bilmiyoruz. Farklı genetik gruplarda daha fazla pik mümkün. Ancak şimdiye kadar vCJD nin diğer dalgaları İle ilgili kanıt yok. vCJD li hastaların periferal dokularında özellikle lenforetiküler doku olmak üzere normal CJD lilerden daha fazla infeksiyöz ajan bulunması nedeni ile kaygılar var. Bu durum cerrahi enstrümanların sterilizasyonu ve iatrojenik yayılım olasılığı olduğuna dair soruları artırmaktadır. Bu aynı zamanda Amerika Birleşik Devletleri kan desteği koruma kaygısı ve konservatif nedenlerinden biridir (İngiltere'de ve Avrupa'da önemli bir zaman geçirenlerin tarama dışına alınması). Birleşik Krallıkta vCJD ajanlarının 2 vakaya kan transfüzyonu aracılığı ile bulaşmış olabileceği olasılığı bulunmakta. Her iki vakada da kan lökosit filtreli değilmiş. Bu durum, lökosit filtrasyonunun kan transfüzyonu aracılığı ile bulaş ihtimalini azaltabileceğini düşündürmüştür.
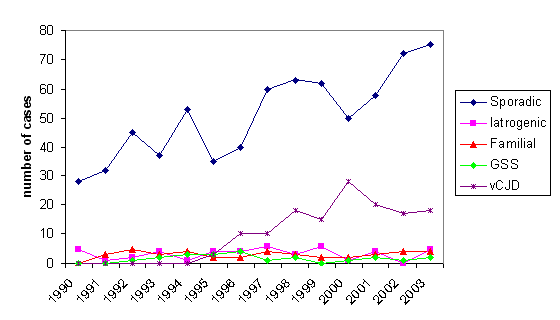
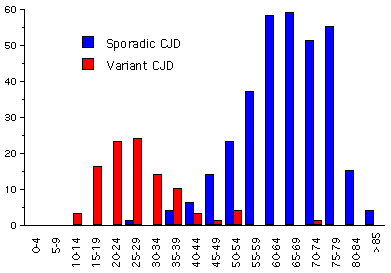 Şekil 8A
Şekil 8A1994-2001 ABD CJD ve vCJD: Birleşik Krallık içinde CJD'nin Yaş Gruplarına Göre Dağılımı
 Şekil 8B
Şekil 8B1990-2003 İngiltere'de CJD nedeniyle ölümlerin sayısı
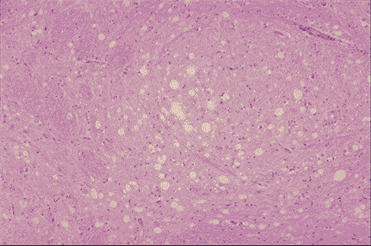
 Şekil 9A
Şekil 9A
BSE'den etkilenen ineğin medullası: Vakuoller nöron ve nötrofillerde görülür.
Küçük çekirdek proliferasyonu ile astrositler. Beyinde inflamatuar hücreler
infiltratı yoktur. Sol x100, Sağ x200
Dr. M. KUBO NIAH, Japan
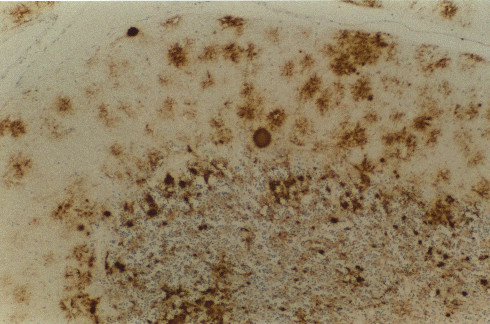 Şekil 9B
Şekil 9B
Beyincikte PrP için immünsitokimya moleküler tabakada bol perisellüler
birikim ve granüler tabakada birden fazla küçük plaklar ile bir kuru-
tip plağın (ortada) güçlü boyanmasını gösterir
© The UK Creutzfeldt-Jakob Disease Surveillance Unit/The Lancet
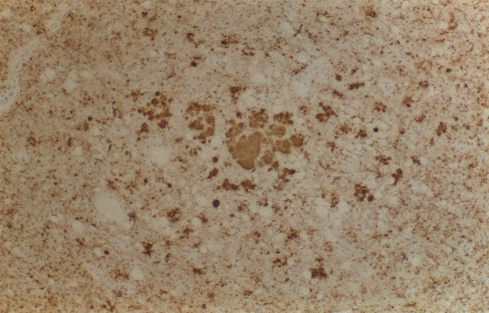 Şekil 9C
Şekil 9C
Talamusta PrP için immünsitokimya çevreleyen nöropilde perivasküler ve
sinaptik birikimi ile birkaç büyük çok merkezli plakları (ortada)
gösterir
© The UK Creutzfeldt-Jakob Disease Surveillance Unit/The Lancet
Gerstmann-Sträussler-Scheinker sendromu
Gerstmann-Sträussler-Scheinker syndrome (GSS) Kuru ya benzeyen semptomlara sahip bir hastalık. Familyal bir hastalık ve sıklıkla CJD vakalarının genetik taşınan bir alt sınıfı olarak kabul edilmekte. Bu hastalık laboratuar hayvanlarına bulaştırılabilmektedir.
Fatal Familial Insomnia
Tedavi edilemeyen progresif bir insomnia, sirkadiyen ritm kaybı, endokrin bozukluklar, motor bozukluklar, demansla sonuçlanan bir insan hastalığıdır. Yine laboratuarda hayvanlara bulaştırılabilen familyal bir hastalıktır. Hastalığın bu formunda, hipotalamus fonksiyonları primer hedef olabileceği görülmektedir.
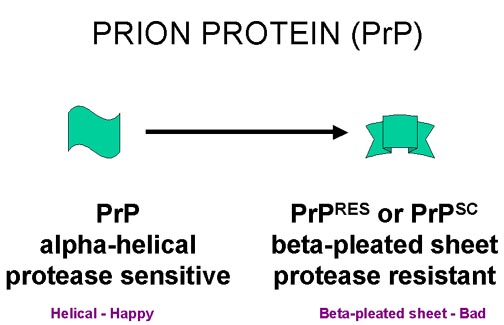
PRİON PROTEİN
İnfekte materyalden hazırlanmış preperatlar, yüksek miktarda PrP içerir (Şekil 9, 10). Bu konak hücre geni tarafından kodlanır ve bir hücre yüzey glikofosfoinozitol(GPI) bağlı proteindir. Normal fonksiyonları bilinmemektedir. İnfekte form, normal formla aynı aminoasit sekansı ve aynı posttranslasyonel modifikasyona sahip, ancak hastalıklı dokudaki konformasyonu farklıdır. Normal form çokça alfa helix içeririrken, hastalıkla ilişkili form çokça beta kıvrımlı tabaka içerir. Hastalıkla ilişkili form daha çok proteaz dirençli olduğundan PrPRES ya da ilk scrapie infeksiyonunda bulunduğundan PrPSC olarak bilinir.
Niçin bir protein infeksiyözdür?
Dirençli formun, normal formdan dirençli forma dönüşmüş olabileceği bir hipotezdir, bu daha sonra dirençli forma daha fazla normal şeklini dönüştürecek; Böylece dönüşüm oranı dirençli formun konsantrasyonu arttıkça giderek kuvvetlenecek. Bu dönüşümün bir kısmından azının ekstrasellüler meydana gelmiş olduğu görünmekte.
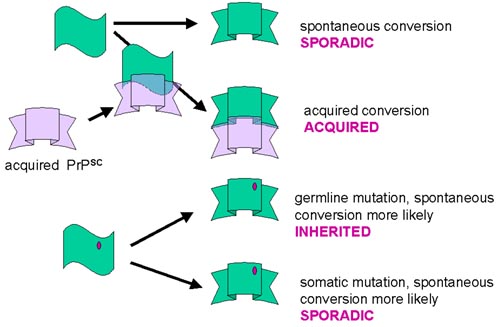
Kazanılmış vakalar
Rezistan formla infekte olma sonucu edinilmiş vakalar olabilir ve bu daha sonra kişinin normal formu PrPSC forma dönüşebilir ve süreç yavaş yavaş yukarıdaki gibi artarak devam edecektir
Sporadik vakalar
Sporadik vakalar normal formun rezistan forma spontan dönüşümünden dolayı olabilir ve bu süreç dirençli forma daha fazla normal form çevrilmesi olarak yükseltilir. Sporadik vakalar rezistan formda spontan dönüşüme uğrayan PrP yapan somatik mutasyonlardan dolayı olabilir veya bilinmeyen bir mekanizma aracılığı ile edinilmiş olabilir.
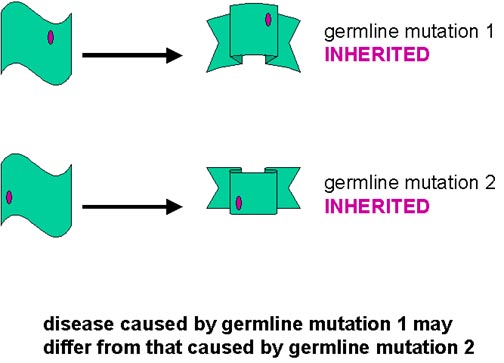
Ailesel (herediter) vakalar
Herediter vakalarda PrP gen mutasyonları izlenmektedir. Bu hastalığın herediter formlarında, mutant formda protein daha büyük olasılıkla rezistan forma spontan değişimle oluşmuş olabilir ve daha sonra aynı birikim süreci ortaya çıkmış olacaktır. Bu mutasyonlardan en az bir kısmında, mutant gene sahip olan herkeste er ya da geç CJD/GSS gelişimi görülür, eğer yeterince uzun yaşarlarsa. Heredite formdaki mutasyonların türü hastalığın klinik seyrini etkileyebilir.
IMMUN CEVAP
Bu sıradışı virüs yada ajanlar bir immün yanıta yol açmazlar.
İnterferon salgılatmazlar. Bu ajanlara karşı antikor yanıtı yoktur. Bu sebeple
bu ajanları ortaya çıkarmak için antikor bakarak insanları taramak mümkün
değildir.
TEDAVİ
Bugune kadar prion hastalıkları değiştirilemeyecek şekilde ölümcüldür. Klasik
CJD genellikle semptomların görülmesinden sonra birkaç ay içinde ölümle
sonuçlanır. Semptomların başlangıcından itibaren ortalama yaklaşık 16 ay içinde
-vCJD de daha uzun- ölüm görülür. CJD hastalarınının bu kötü prognozundan dolayı
çeşitli ilaçlar etkinlik için test edilmiş ancak şimdiye kadar eğer pozitif
etkileri varsa da az talep görmüşlerdir. Ve pozitif etki vardıysa da çok kısa
süreli idi. Üstelik bu ilaçların yan etkileri çok ciddi olabilir.
Diğer yaklaşım faredeki prion formasyonlarını inhibe eden
antikorlar yapılması. Heyecan verici kısmı şu ki eğer birden daha fazla PrPSC
formasyonu durdurulursa, hücreler aslında var olan PrPSC leri yıkabilir
görünmekte. Bu yaklaşım henüz insanlar üzerinde denenmemiş.
Bu yanını hatırla ki eğer progresyonu yavaşlatan ilaçlar var ise, semptomları
olan birilerini tedavi edemeseler de, semptom gelişmemiş familyal formlarda
kullanılabilirler. Bu prion hastalıklarında yeni bir alan.
TANI
Yaşam boyunca, olası tanı klinik tabloya dayanır. EEG bazı vakalarda işe yarar destekleyici bulgu sağlar. Semptomların geniş bir kısmı ve hastalık seyri tanıyı zorlaştırır ve prion hastalıkları sıklıkla atlanır. Kesin tanı sıklıkla post mortem beyin örnekleklemesi ile yapılır. Beyin biyopisisi kullanılabilir değil. Seroloji hastaların immün yanıt göstermemeleri nedeni ile kullanılamamaktadır.
vCJD vakalarında MRI tanı için kullanılabilir. Periferik lenfoid
dokunun PrPSC bulundurmasından dolayı örneğin tonsil biyopsileri uygulanabilir
pozitif bir bulgu olabilir. PrP gen delesyonuna sahip fareler kullanılarak PrPSC
antikorları yapmak mümkün ve dahası proteine tolerans kazandırılmamşıtır. Eğer
hastadan yeterli prion materyali alınabilirse bu antikorlar western blot
testlerinde kullanılabilir.
 Şekil 10
Şekil 10Prion proteini PrP (hücresel bir gen tarafından kodlanır ve normal hücrelerde yapılır) iki formda bulunabilir. Hastalıklı dokuda bir çok beta-kıvrımlı bir tabakada proteaza dirençli bir form (PrPSC) 'amiloid plaklar "olarak birikir
Prion hastalıkları için basitleştirilmiş model.
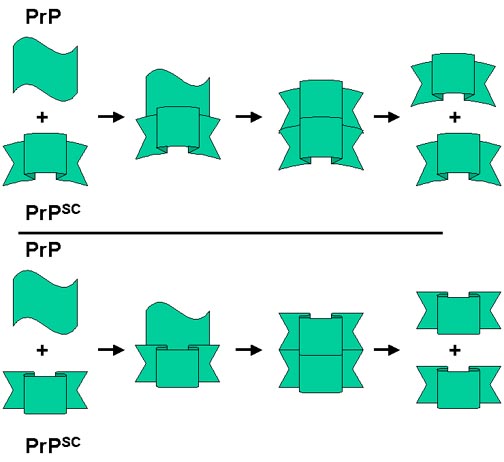
PrPSC, proteaz dirençli molekül, bir taslak gibi davranır. Helikal formun sonradan dirençli formdaki beta kırmalı tabakaya dönüşümüyle ilişkilidir (Muhtemelen normalde bu durumu önlemek enerji engelleri azaltarak). Rezistan formun bir şablonu gibi davranan ve bu süreci hızlandıran iki molekül var.
Bu model nasıl sporadik, edinilmiş ya da herediter formları açıklayabilir? Alfa helikal formdan beta kıvrımlı tabakaya dönüşüm çok nadir (sporadik) olmasına rağmen spontan meydana gelebilir. Bu dönüşüm bazı ekzojenöz kaynaklardan(edinilmiş) gelen PrPsc tarafından katalizleniyor olabilir. Germ line mutasyonları daha olası şekilde spontan dönüşüm yapabilir(kalıtımsal). Somatik mutasyonlar muhtemelen spontan olarak dönüşüm yapabilir (sporadik). Bu durumda PrPsc molükülü ile sonuçlanan mutant forma dönüşüm süreci başlayabilir ve sonra çevredeki hücrelerden normal fom çevrimi olur
Prion hastalıklarında niçin farklar var? İçerilen mutasyon ya da PrPsc kaynağına göre prion proteinin proteaz dirençli formlarında ince farklar var olabilir. Şekilde görüldüğü gibi, PrPsc nin birbiriyle ilişkili ancak küçük miktarda farklı 2 formu normal formdan kendi konformasyonlarına dönerler. Böylelikle final PrPsc ürünü başlangıç sürecindeki forma bağlı olarak birikir.
Bu açıklama heredite formlarda uygulanabilir. Farklı mutasyonlar, farklı proteaz dirençli formların spontan oluştuğu kabul edilerek, PrPsc ye predispozan olabilir
BULAŞICI ENSEFALOPATİLER VE DİĞER HASTALIKLAR
Amiloid plaklar diğer SSS hastalıklarında da görülür. Ancak Kuru, CJD ve GSS de görülen bu amiloid plakların büyük komponenti örneğin Alzheimer hastalığında görülenlerle aynı materyalden değildir. Amiloidi işaret eden boyanma özellikleri birçok glikozile protein agregatlarının boyanma özellikleri ile aynı olabilir.
Prion hastalıklarının SSS’de hücrelerin fonksiyonlarıyla ilgili
karışıklıklar oluşturması, SSS’deki sinir dokusunun progresif dejenerasyonuna
yol açan bozuklukların en önemli noktası olması mümkün. Prion hastalıklarının
patogenezini anlamak SSS nin diğer hastalıklarının anlaşılmasına yardımcı
olabilir.
Mikrobiyoloji ve İmmünoloji On-line, Viroloji Bölümüne Dönünüz
This page last changed on Wednesday, November 23, 2016
Page maintained by Richard Hunt