|
x |
x |
|
 |
 |
|
INFECTIOUS
DISEASE |
BACTERIOLOGY |
IMMUNOLOGY |
MYCOLOGY |
PARASITOLOGY |
VIROLOGY |
|
|
VIROLOGY - CHAPTER NINE
ANTI-VIRAL CHEMOTHERAPY
Dr Richard Hunt
Professor
University of South Carolina School of Medicine
Columbia
South Carolina
|
|
En
Español |
|
SHQIP - ALBANIAN |
|
TURKISH |
Let us know what you think
FEEDBACK |
|
SEARCH |

 |
|
|
|
TEACHING
OBJECTIVES
To elucidate the drugs that are currently used as anti-viral agents and
to determine why they are effective agents. The mode to action of these drugs will be
discussed |

|
|
SEE ALSO
Anti-HIV Chemotherapy chapter |
Anti-bacterial drugs such as the penicillin antibiotics have proved very successful
since they act against a bacterial structure, the cell wall, that is not present in
eukaryotic cells. In contrast, most anti-viral agents have proved of little use
therapeutically since the virus uses host-cell metabolic reactions and thus,
for the most part, anti-viral agents will also be anti-cell agents. Thus, the alternative
approach of stimulating the host's immune responses using vaccines has been most
often pursued. Nevertheless, there are activities (i.e. enzymes) that are virus-encoded
and therefore offer potential virus-specific targets. This is particularly
the case with the viruses that have large genomes and code for their own replication
enzymes. Even so, unfortunately, many anti-virals that are apparently effective in vitro
are ineffective in vivo.
A successful anti-viral drug should:
(i) interfere with a virus-specific function (either because the function is unique to
the virus or the similar host function is much less susceptible to the drug)
or
(ii) interfere with a cellular function so that the virus cannot replicate. To
be specific, the anti-viral drug must only kill virus-infected cells. This could be done by restricting drug
activation to virus-infected cells.
An ideal drug should be:
- Water-soluble
- Stable in the blood stream
- Easily taken up by cells
An ideal drug should NOT be:
- Toxic
- Carcinogenic
- Allergenic
- Mutagenic
- Teratogenic
Toxicity of an anti-viral drug may be acceptable if there is no alternative:
such as, for example, in symptomatic rabies or
hemorrhagic fever
Obviously, a good drug must show much more toxicity to the virus than the host cell.
We measure selectivity by the therapeutic index of the drug
Therapeutic index (T.I.): Minimum dose that is toxic to cell
Minimum dose that is toxic to virus
Effective drugs have a T.I. of 100-1000 or better.
Just as with anti-bacterials, we must find a virus Achilles heel. This could be
an enzyme that is unique to the virus so that the drug is not toxic to the host cell.
The following is a list of viruses that are known to code for their own enzymes. Among
the other enzymes are: proteases, mRNA capping enzymes, neuraminidases,
ribonucleases, kinases and uncoating enzymes.
|
|
Molecular Structure pop-up boxes
show chemical and three-dimensional structures
|
| |
| Virus |
RNA/DNA polymerase |
Other |
| Picorna |
+ |
+ |
| Reo |
+ |
+ |
| Toga |
+ |
+ |
| Orthomyxo |
+ |
+ |
| Paramyxo |
+ |
+ |
| Rhabdo |
+ |
+ |
| Arena |
+ |
? |
| Corona |
+ |
+ |
| Bunya |
+ |
? |
| Parvo |
- |
+ |
| Adeno |
+ |
+ |
| Herpes |
+ |
+ |
| Irido |
+ |
+ |
| Pox |
+ |
+ |
| Hepatitis B |
+ |
+ |
|
| |
The very first licensed anti-viral drug was idoxuridine (1963), a pyrimidine
analog that inhibits viral DNA synthesis. It is still used topically for
epithelial herpetic keratitis but has largely been replaced because other drugs
are less toxic. It is toxic because it lacks specificity, i.e. the drug inhibits host DNA
polymerization as well as that of the virus.
One of the better anti-viral drugs that we have dates from 1983: Acyclovir
(acycloguanosine) which is a purine analog. It inhibits herpes DNA replication.
It is also a nucleoside analog but, in contrast to idoxuridine, is highly specific and
does not exhibit severe toxic side effects...for the reason for this, see below.
|
 Figure 1
Figure 1
Cellular targets for drugs
|
POSSIBLE PHASES OF LIFE CYCLE ON WHICH ANTI-VIRAL ATTACK MIGHT BE LAUNCHED
The life cycle of a virus comprises several stages such as binding to
the cell surface, replication, protein synthesis etc. and all of these
stages may be the target of anti-viral drugs. Among the life cycle
stages that have been targeted by potential therapeutic agents are:
- Attachment of the virus to the cell surface, perhaps
as a result of competition with a specific viral receptor.
- Uptake into intracellular vesicles (endosomes)
- Uncoating of virus (loss of protein coat, fusion of lipid membrane with
endosome/lysosome). Note: the endosome/lysosome compartment is acidic and inhibition of
acidification of this compartment might be a good target.
- Integration of the viral DNA into
chromosomal DNA of the host cell (where this occurs).
- Transcription of genome to new RNA or DNA (polymerases are the target).
- mRNA transcription
- mRNA processing (poly adenylation, methylation, capping, splicing)
- Translation to protein
- Post-translational modification of proteins (glycosylation, phosphorylation, fatty acylation, proteolysis). Some of these are essential for functional, infective viral
progeny.
- Assembly of the components into the whole virus
We shall look at each of these life-cycle stages (figure 1) in the
following sections.
|
|
WEB RESOURCES
Classes
of anti-HIV drugs
NSAID |
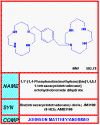 Figure 1a
Figure 1a
AMD3100 |
BINDING TO RECEPTOR OR UPTAKE INTO INTRACELLULAR VESICLES
There, were until recently, no good drugs that stop
receptor binding by any virus (but see
influenza sialidase inhibitor below). However, possibilities include the use a peptide that mimics the receptor such
as soluble CD4 protein. This would bind HIV gp120 and stop it binding to the
receptor on the cell surface. However, there is a stability problem. The soluble protein is
rapidly broken down and cleared from the circulation, i.e. an efficacious concentration is
not achieved for a useful period. Attempts have been made to stabilize proteins but little
success has been achieved. There have been attempts to couple soluble CD4 to toxins to
kill infected cells, again with little success. In some cases, soluble CD4 can
make the virus more infectious in laboratory studies. It is not known why this
happens but a possible explanation might be that binding to gp120 causes a
conformational change in the latter giving it a higher affinity for the
co-receptor that is important, along with CD4 antigen, in infection of a cell by
HIV (see HIV, section 7). It is also possible that soluble CD4 bound to gp120 might promote
fusion.
PRO 542 is a tetrameric form of soluble CD4 antigen genetically fused to an
immunoglobulin for added stability. This CD4-immunoglobulin fusion protein comprises the D1
and D2 domains of human CD4 and the heavy and light chain constant regions of
human IgG2. It has a high affinity for gp120.
For HIV to infect a cell, it must bind both to CD4 antigen and to a
co-receptor, a chemokine receptor. The chemokine receptors bind chemokines and
these can block binding to HIV gp120. Derivatives of one such chemokine (RANTES)
have been used as agents to block virus binding. In addition to binding to the
CCR5 chemokine receptor, these derivatives, like the natural chemokine,
down-regulate the co-receptor by endocytosis, making it more difficult for the
virus to bind. Chemokines such as RANTES are pro-inflammatory and chemotactic
for leukocytes but these properties can be reduced by chemical modification at
the N-terminus. Such chemokine derivatives are excellent antagonists of HIV
binding and can protect monkeys that are exposed to HIV in the vagina.
Anti-co-receptor monoclonal antibodies are also being developed to block virus
binding. Another approach is to use peptides that are analogous to the
transmembrane sequence of the co-receptor; these disrupt the interaction between
the seven transmembrane alpha helices of the co-receptor protein.
AMD-3100
Chemical name: 1,4,8,11-Tetraazacyclotetradecane,
1,1'-(1,4-phenylenebis(methylene))bis-, octahydrochloride
In addition to peptide approaches to disrupt HIV-co-receptor interactions, some small
molecule inhibitors have been developed. For example, AMD3100/JM-3100 appears to
bind to the ligand binding site of the co-receptor known as CXCR4 (fusin) and blocks the interaction
between CXCR4 and the V3 loop of gp120.
Maraviroc
Chemical name:
4,4-difluoro-N-{(1S)-3-[exo-3-(3-isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1-phenylpropyl}cyclohexanecarboxamide.
Maraviroc (brand-named Selzentry, or Celsentri outside the U.S.) was approved
for use in HIV-infected patients in August 2007. It blocks the interaction
between chemokine receptor CCR5 and HIV gp120. Because HIV can also use another
co-receptor, CXCR4, an HIV tropism test is performed to determine if the drug
will be effective. In a study comparing Maraviroc plus the conventional HAART
triple combination of drugs with the standard of care HAART alone, use of HAART
plus Maraviroc gave twice as many patients with HIV levels of fewer than 50
copies/ml compared to standard HAART.
FUSION OF VIRAL AND HOST CELL MEMBRANE
Agents
that block fusion of HIV with the host cell by interacting with gp41
Enfuvirtide
Other names: DP-178, pentafuside, T-20, Fuzeon®.
Peptides derived from gp41 can inhibit
infection, probably by blocking the interaction of gp41 with cell membrane
proteins during fusion or by stopping the conformational change that results
from the association of two gp41 molecules and which is necessary for fusion. Enfuvirtide
(Fuzeon) is a 36 amino acid peptide that corresponds to residues 127-162 of
gp41 and blocks this conformational change. In clinical trials, a nearly two log
reduction in plasma viral levels was achieved. This drug was approved in 2003
but recent reports suggest low bioavailability and the emergence of resistant
mutants.
There is a cavity on gp41 that could
hold a small molecule inhibitor. Peptides containing D-amino acids that would
fit this cavity have been identified and inhibit fusion.
Others
RFI-641
Chemical Name:
4,4"-bis-{4,6-bis-[3-(bis-carbamoylmethyl-sulfamoyl)-phenylamino]-(1,3,5)
triazin-2-ylamino}-biphenyl-2,2"-disulfonic acid
RFI-641 (biphenyl triazine) inhibits fusion of the
membrane of respiratory syncytial virus (RSV) with the cell membrane. It seems
to alter the conformation of the fusion (F) protein of the virus and is active
in
vivo in several animal models. It is active against RSV A and B strains. The drug is much
better than ribovirin (the only routinely used drug in treating RSV infections)
and seems to be RSV-specific. The drug has now been abandoned for routine use
because of toxicity problems and delivery problems. It cannot be taken orally
and so is delivered as an aerosol but elderly patients would likely find such a
mode of delivery problematical. It may be of use in infants and derivatives may
be less toxic.
BMS-433771
Chemical Name: 2H-Imidazo(4,5-c)pyridin-2-one,
1-cyclopropyl-1,3-dihydro-3-((1-(3-hydroxypropyl)-1H-benzimidazol-2-yl)methyl)-
BMS-433771 is an RSV fusion inhibitor. It works by inhibition of viral F
protein-induced membrane fusion and is active against both A and B
groups of RSV. It is efficacious against RSV infection in two
rodent models when dosed orally prior to infection and may be of
clinical use.
|
 Figure 2
Figure 2
Arildone |
UNCOATING AND ENTRY INTO THE
CYTOPLASM
Uncoating of the virus (i.e. the loss of the lipid
envelope of
membrane-containing viruses or the loss of nucleocapsid proteins in
non-enveloped viruses)
often occurs in low pH endosome or lysosomes, as the result of a pH-dependent fusogen.
Note: Some viruses do not need an acidic environment for fusion and fuse with the plasma membrane; this is the case with
herpes viruses and HIV and leads to the formation of
syncytia.
|
 Figure 3
Figure 3
Human rhinovirus with WIN V1 (arrows) buried in a pocket in the VP1
protein
|
Arildone
and the WIN compounds
Chemical name: 4-(6-(2-Chloro-4-methoxy)phenoxy)hexyl-3,5-heptanedione
Arildone and the WIN compounds inhibit uncoating of picornaviruses,
which do not have a lipid membrane. The drug inserts
into a canyon in VPI protein of virus and blocks ion transport.
For more information see
chapter 10, part 3.
Pleconaril
Chemical name:
3-(3,5-Dimethyl-4-(3-(3-methyl-5-isoxazolyl)propoxy)phenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole
Other names: Win 63843, Picovir
This acts like a WIN compound in that it fits
into a hydrophobic pocket in the nucleocapsid and interrupts the replication of
the virus by stopping the shedding of nucleocapsid proteins from the RNA. This
orally taken compound is broadly active against a variety of entero- and
rhinoviruses (picornaviruses) but the reduction in the duration of symptoms is
small and only occurs in some populations. An intranasal formulation of pleconaril
represents an optimized delivery approach, as compared to the earlier oral
formulation.
|
|
MOLECULAR STRUCTURE
Arildone
Pleconaril |
 Figure 4
Figure 4
Amantadine (left) Rimanadine (right)
Figure 4a
Hydroxychloroquine
This drug has an extra hydroxyl group compared to chloroquine
|
Amantadine
Chemical name: tricyclo[3.3.1.1.3,7]decane-1-amine hydrochloride
Other names: 1-adamantanamine, amantadine HCl, Symmetrel®,
Mantadix®, Amantan®
Rimantadine
Chemical name: alpha-methyltricyclo[3.3.1.1.3,7]decane-1-methanamine
hydrochloride
Other names: alpha-methyl-1-adamantanemethylamine HCl, Flumadine®
These were originally thought to be lysosomotropic,
that is they were thought
to stop acidification of the endocytic vesicles and lysosomes. However, they are
now known to act on a viral protein,
the M2 ion channel, which is necessary for the acidification of the enveloped
virus in the endosome, a process that must occur
before uncoating of the virus. These
drugs may also act on maturation of influenza HA glycoprotein so that progeny
virions are
poorly infective.
These drugs are good for oral prophylaxis against influenza A (but not influenza B). They
are a good alternative to the vaccine in immunocompromised patients and the elderly. Other
than this, they are not used much in western countries. Prophylactic rimantadine has been
used a lot in countries of the former USSR. Both of these drugs are licensed for use in
US. Interest in these drugs has rearisen because of the possibility of an avian
flu pandemic since currently there is no vaccine for this type of influenza
virus (H5N1) and it will take several months to develop a vaccine after the
pandemic strain is identified.
In the 2005-2006 influenza season, 92% of
H3N2 strains examined had a mutations that would confer resistance to these
drugs as did 25% of the H1N1 strains tested. Similar problems were seen in
2006-2007 and so these drugs are not recommended until the per cent resistance
in the major circulating types drops.
Chloroquine and hydroxychloroquine
Figure 4a
Chloroquine and hydroxychloroquine are widely used as anti-malarial drugs
and also for the treatment of rheumatoid arthritis, lupus, and porphyria
cutanea tarda. In the spring of 2020, a now retracted paper suggested the
use of hydroxychloroquine in combination with the macrolide antibiotic
azithromycin as a treatment for patients infected with SARS-CoV-2.
Experiments in cultured Vero cells showed that hydroxychloroquine was more
potent than chloroquine at inhibiting SARS-CoV-2 in vitro with a high
selectivity index; as a result, numerous clinical trials of these old drugs
alone or in combination with other drugs are being conducted. However,
previous attempts to use them as anti-virals in vivo have shown mixed
results and sometimes they have exacerbated the disease. Chloroquine
inhibited Ebola virus replication in vitro but caused rapid worsening of
Ebola infection in guinea pigs and made no difference to mortality in mice
and hamsters. Similarly, these drugs worsened symptoms in chikungunya virus
infections in monkeys. In April 2020, the first results of a trial of
hydroxychloroquine showed that in a group of US veterans treated for
COVOD-19, the risk of death from any cause was significantly greater in
those receiving the drug compared to those who did not. In addition, a
randomized trial in China showed no evidence of the efficacy of
hydroxychloroquine.
Thus, what seem to be promising results in vitro (i.e. cell culture) are
often not reflected in vivo. It should also be noted that while these drugs
have been used clinically for a long time, especially as anti-malarials,
they are not without serious side effects including inducing cardiac
arrhythmias in genetically predisposed patients in whom the drugs block ion
channels that are involved in heart beat control. Azithromycin was probably
used in the original study to control bacterial infections and it can
increase cardiovascular death when used in association with
hydroxychloroquine in rheumatoid arthritis patients.
Although the chloroquines (4-aminoquinolines) have long been used as
anti-malarials, their mechanism of action is still poorly understood. They
are lysosomotropic agents that inhibit the acidification of late endosomes
and lysosomes and may block entry of some viruses into the cytoplasm. This
has been suggested for their effects on HIV replication. In the case of some
coronaviruses and other viruses, an acidic environment is required for a
conformational change in the virus surface protein (the S protein in the
case of coronaviruses) so that the fusogen region of this molecule can
promote the fusion of the viral envelope membrane with the endosomal/lysosomal
membrane resulting in entry of the nucleocapsid into the cytoplasm. Another
possibility suggested by molecular modeling, is that both drugs can bind
sialic acid and gangliosides with high affinity. A ganglioside binding site
was also identified on the SARS-CoV-2 S protein which may be involved in the
attachment of the virus to lipid rafts in the cell plasma membrane as a
preliminary to the attachment of the S protein to the ACE-2 receptor. Thus,
it may be that the drugs, by binding gangliosides, inhibit the binding of
the virus to cell surface receptors.
|
|
|
|
MOLECULAR STRUCTURE
Amantadine
Rimantadine |
 Figure 5
Figure 5
Acyclovir is
phosphorylated first by a viral kinase to acycloGMP and
then by cellular kinases to acycloGDP and acyclo GTP
|
NUCLEIC ACID SYNTHESIS
The best anti-viral drugs that we have are of this type.
They are selective because:
- the virus may use its own enzyme to activate
the drug
and/or
- the viral polymerases may be much more sensitive to the drug than the corresponding
host enzymes
|
 Figure 6
Figure 6
Three phosphates are added to thymidine. The first is added by the viral
enzyme and the remainder by cellular enzymes
|
Thymidine kinase substrates
The thymidine kinase (figure 6) of herpes simplex (and other) viruses allows the
virus to grow in cells that do not have a high concentration of phosphorylated nucleic
acid precursors. These are usually cells that are not replicating their genome (e.g. nerve cells). Resting cells do,
however, have
unphosphorylated nucleosides. By bringing in its own kinase, the virus can grow in
non-dividing cells by phosphorylating the cells' nucleosides.
The name of the enzyme is a bit of a misnomer since it can work on other nucleosides
than thymidine (thymidine happens to be the best substrate), i.e. the enzyme is
non-specific as to substrate. This is in contrast to the host cell thymidine kinase which
is very specific to thymidine since the cell has other enzymes to phosphorylate
the other nucleosides. This lack of specificity of the viral enzyme allows
it also to work on nucleoside-analog drugs and phosphorylate them. The host enzyme,
because of its greater specificity, is much less good at this (and often does not
phosphorylate the drug at all).
The fact that the viral enzyme is quite good at phosphorylating the drug has another
advantage. We can administer the nucleoside-analog in a non-phosphorylated form. This is
useful as it is difficult to get phosphorylated drug into the cell because plasma
membranes are poorly permeable to phosphorylated compounds in the absence of a specific
transport protein.
Thus the need for activation restricts use of drug to viruses with their own
thymidine kinase or
that cause cell to overproduce the endogenous enzyme (which may, if we are
lucky, activate the drug to a lesser degree).
To recapitulate, the great use of these drugs results from the facts that:
- they are only activated by the virus-infected cell
- the activated form of the drug is rendered even more specific as a result of the
viral DNA polymerase being more sensitive to the drug than the host enzyme.
Most nucleic acid synthesis inhibiting drugs are nucleoside analogs with
an altered
sugar, base or both. Acyclovir (acycloguanosine) is the best example of such a drug
and is used to treat herpes virus infections. It gets into the cell across the
plasma membrane as the nucleoside form and is
then specifically phosphorylated inside the cell by herpes virus thymidine kinase to an active form.
It
then blocks DNA synthesis by inhibiting polymerization; it is a chain
terminator.
|
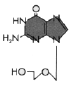 Figure 7
Figure 7
Acyclovir
|
DNA Synthesis Inhibitors
Sugar modifications
Acyclovir/Acycloguanosine
Chemical name:
9-(2-hydroxyethoxymethyl)guanine, acycloguanosine (ACG)
Other names: Aciclovir (ACV), Zovirax®.
(figure 7).
As noted above, this drug is very
selective and one of our better anti-viral drugs. It is non-toxic to uninfected cells (except
some renal dysfunction) because it is not activated by uninfected cells (because
the drug is a poor substrate for the very specific cell thymidine
kinase). Moreover, the DNA polymerase of
herpes simplex virus is 10 times more
sensitive than cellular DNA polymerase. This drug is a competitive inhibitor -
it competes with dGTP - but it also acts in another way that is more important:
When it gets incorporated into DNA, it acts as a chain terminator (figure 8). It
is taken orally, topically or intra-venously.
HSV-1, HSV-2 and VZV
are susceptible to acyclovir.
|
 Figure 8 Figure 8
Chain termination
|
Acyclovir is effective against herpes simplex
keratitis, latent HSV, fever blisters (H. labialis),
genital herpes. Acyclovir-resistant mutants are a problem after long term use and
have been shown to result from changes in the thymidine kinase or polymerase gene.
There is a
prodrug form of acyclovir called Valaciclovir ((VACV), Zelitrex®,
Valtrex®) which is an L- valine ester of the drug. This can be taken
orally.
Penciclovir
Chemical Name: 9-(4-hydroxy-3-hydroxymethyl-but-1-yl)guanine
Other names: PCV, Denavir®, Vectavir®
Used against HSV-1 and -2 and VZV, Penciclovir is similar in action to
acyclovir, that is it is a chain terminator. It can only be used as a topical
cream because of insolubility.
Famciclovir
Chemical Name: diacetyl ester of
9-(4-hydroxy-3-hydroxymethyl-but-1-yl)-6-deoxyguanine
Other names: FCV, Famvir®.
This is a
prodrug
of Penciclovir and is converted to Penciclovir as a result of oxidation and the
hydrolysis of the two ester groups. Because of the esterification, it is soluble
in water and can be administered orally. It is also used for HSV-1 and -2 and
VZV infections.
|
|
MOLECULAR STRUCTURE
Acyclovir |
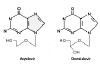 Figure 9
Figure 9
Acyclovir Gancyclovir
|
Ganciclovir
Chemical name:
9-(1,3-dihydroxy-2-propoxymethyl)guanine
Other names: DHPG, GCV, Cymevene®, Cytovene®
- figure 9
This drug is very similar to Acyclovir, it just has an
extra -OH. It is also available as a
prodrug
called Valganciclovir which is an L-valine ester of Ganciclovir (Valcyte).
Oral Valganciclovir will probably to replace intravenous Ganciclovir for
therapy and prevention of cytomegalovirus
(CMV) infections. Ganciclovir is active against CMV for which it is the drug of choice. Acyclovir has
some activity against CMV in culture but has not found much use in
therapy of these infections because of the superiority of Ganciclovir.
As with Acyclovir, Ganciclovir targets the viral DNA polymerase and
acts as a chain terminator. In herpes virus-infected cells, it is
phosphorylated first by the viral thymidine kinase and then by cell
kinases to yield the triphospho form of the drug which is incorporated
into and terminates the DNA chain. However, CMV does not encode a thymidine
kinase.
Instead, Ganciclovir is phosphorylated by a CMV-encoded
protein kinase (UL97) which accounts for its specificity for infected
cells. Selectivity is also achieved because the viral polymerase has 30 times
greater affinity for Ganciclovir than the host enzyme.
Normally, Ganciclovir is given intra-venously at a level of 10mg/kg
per day or orally at 3000mg/day. It is often used for CMV retinitis in AIDS
patients for whom there is an intraocular (that is, intravitreal) implant known
as Vitrasert. This contains 4.5 mg Ganciclovir for localized therapy.
|
|
MOLECULAR STRUCTURE
Ganciclovir |
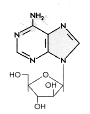 Figure 10
Figure 10
Ara-A
|
Adenosine arabinoside
Chemical name:
9-beta-D-Arabinofuranosyl-9H-purin-6-amine
Other names: Vidarabine,
Ara-A - figure 10
Acyclovir and Ganciclovir are chain terminators because they do not have a
complete sugar ring; the appropriate 3' -OH group needed to form a phosphodiester bond during DNA elongation
is missing. Adenosine arabinoside has a
complete sugar but it is arabinose rather than ribose. This drug has severe side effects and is only used in
potentially lethal disease. In addition, it is easily deaminated in the bloodstream to a less
effective form, ara-hypoxanthine
|
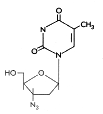 Figure 11A Figure 11A
AZT
|
Zidovudine
Chemical name:
3′-azido-2′,3′-dideoxythymidine
Other names: Azidothymidine, AZT, Retrovir® - figure 11A
This drug is also a chain
terminator. It is phosphorylated by a cell kinase and so it can be used
against viruses without their own thymidine kinase (e.g. HIV). Reverse transcriptase
(RNA-dependent DNA polymerase) is more sensitive to the drug than human
DNA-dependent DNA polymerase accounting for the specificity but there
are severe toxicity effects. It is used as an anti-HIV type 1 and type 2
drug (see HIV). Because of the use
of RNA polymerase II in the synthesis of the viral genome of
retroviruses and the
consequent high rate of mutation of the virus, the selective pressure of
the presence of the drug rapidly leads to the emergence of resistant
viral mutants. All of these have mutations in reverse transcriptase.
Because of the emergence of resistant mutants, AZT is administered in
combination with other drugs.
|
|
MOLECULAR STRUCTURE
AZT |
|
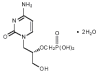 Figure 11B
Figure 11B
Cidofovir
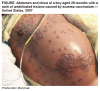 Figure 11C
Figure 11C
Eczema vaccinatum in a 28 month old boy |
Cidofovir
Chemical name: 1-[( S )-3-hydroxy-2-(phosphonomethoxy)propyl]cytosine dihydrate (HPMPC)
Other names: Vistide® - figure 11B
Cidofovir is both a DNA chain terminator and DNA polymerase inhibitor.
It is an acyclic nucleoside phosphonate (not a phosphate) in which the
C-O-P bond in a nucleoside monophosphate has been replaced by a
phosphonate (C-P) bond that provides an enzymatically stable derivative
with a long half life.The drug is administered in the
phosphonomethoxy-nucleoside form and is phosphorylated twice
intracellularly to the active diphosphate form using two cellular
kinases (pyrimidine nucleoside monophosphate kinase and pyrimidine
nucleoside diphosphate kinase. A viral kinase is not involved, in
contrast to acyclovir which is administered as the nucleoside form and
the first phosphate is added by viral thymidine kinase).
Cidofovir inhibits the DNA polymerases of a number of viruses at
concentrations that are substantially lower than those needed to inhibit
human DNA polymerases. It is active against herpes viruses with fewer
side effects than Ganciclovir although it does show nephrocytotoxicity
and a number of other side effects. It must be administered along
with probenecid in order to block renal tubular secretion of the drug.
Cidofovir is particularly useful in the treatment of cytomegalovirus and
is indicated for the treatment of CMV retinitis in patients with
AIDS. It may be useful for treatment of acyclovir-resistant herpes
infections. It is also active against pox viruses, including the molluscum contagiosum virus,
BK virus, which is a polyoma virus,
and adenoviruses. It is promising for the treatment of immunocompromised
patients for gasteroenteritis caused by adenovirus, although no control
studies have been carried out, and has been used as an adjunctive
treatment in addition to HAART in the treatment of AIDS patients with
progressive multifocal leukoencephalopathy (PML). The latter is caused
by JC, another human polyoma virus.
Cidofovir was recently (March 2007) used (along with an experimental
drug, ST-246) in treating a case of eczema vaccinatum in a
two-year old boy. This is an unusual side effect of smallpox vaccination
in which the live vaccinia virus in the vaccine can be passed to
contacts of the vaccinee who are usually immunocompromised. It this
case, because of the eczema, the virus was able to enter the patient's
skin cells and replicate, initially causing a widespread rash and
then blisters with a central dimple which is indicative of vaccinia
infection. The rash encompassed 50% of the patient's keratinized skin.
Although eczema vaccinatum can be fatal, the patient was
discharged after 48 days in hospital.
|
|
|
|
CASE REPORT
Household Transmission
of Vaccinia Virus from Contact with a Military Smallpox Vaccinee |
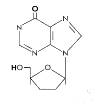 Figure 12
Figure 12
DDI
|
Other sugar modifications
Dideoxyinosine
Chemical name: 2′,3′-dideoxyinosine
Other names: DDI,
Didanosine, Videx®
- figure 12
This is licensed for use against HIV in AZT-resistant
patients and in combination drug treatments along with AZT.
|
|
MOLECULAR STRUCTURE
DDI |
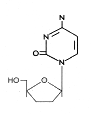 Figure 13
Figure 13
DDC
|
Zalcitabine
Chemical name: 2′,3′-dideoxycytidine
Other names: Dideoxycytosine, DDC, Hivid®, - figure 13
DDC is also licensed for use with AZT in HIV patients.
Again, as with AZT, there is pronounced toxicity because of lack of
specificity to the viral polymerase and the rapid emergence
of resistant HIV mutant strains.Stavudine
Chemical name: 2′,3′-didehydro-2′,3′-dideoxythymidine
Other names: d4T, Zerit®.
This is also used in combination therapy, particularly in advanced
HIV disease.
Lamivudine
Chemical name: (−)-β-L-3′-thia-2′,3′-dideoxycytidine
Other names: 3TC, Epivir®,
Zeffix®).
This is active against HIV types 1 and 2 and also against hepatitis
B virus. In both cases it acts as a chain termination during reverse
transcription. For HIV, 3TC can be administered with AZT in a
combination drug (Combivir®) or with AZT and Abacavir (Trizivir®).
Abacavir
Chemical name:
(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
succinate
Other names: ABC, Ziagen®
Emtricitabine
Chemical name:
(−)-β-L-3′-thia-2′,3′-dideoxy-5-fluorocytidine
Other names: (−)-FTC, Emtriva®.
This is another reverse transcriptase inhibitor that is active
against HIV and hepatitis B virus.
Tenofovir disoproxil
Chemical name: Fumarate salt of bis(isopropoxycarbonyloxymethyl) ester of (R)-9-(2-phosphonylmethoxypropyl)adenine
Other names: bis(POC)PMPA, Viread®
Tenofovir is active against retroviral and hepatitis B reverse
transcriptase and is a chain terminator. It is often used in
combination with lamivudine and a non-nucleoside reverse
transcriptase inhibitor, efavirrenz. It should not be used in
combination with lamivudine and abacavir. In addition to being
licensed for use in treating HIV infection, tenofovir is also
approved for treating hepatitis B.
|
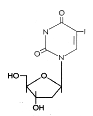 Figure 14
Figure 14
IDU
|
Base modifications
These are pyrimidine analogs that are incorporated into DNA by the viral
DNA polymerase. They form unstable base pairs and mis-translation
results in mutant
proteins. They are competitive inhibitors of the viral DNA polymerase after
intracellular phosphorylation.
Bromovinyl deoxyuridine (Brivudin)
Chemical name: (E)-5-(2-bromovinyl)-2′-deoxyuridine,
bromovinyldeoxyuridine
Other names: BVDU, Zostex®, Zonavir®,
Zerpex®.
BVDU is used for treating HSV (type 1) and VZV. The drug is initially
phosphoryalted by viral thymidine kinase, hence its specificity. It is
used various HSV and VSV infections including HSV keratitis and genital
herpes. It can be given orally or topically.
Iodo-deoxyuridine (Idoxuridine)
Chemical name: 5-iodo-2′-deoxyuridine
Other names: IDU, IUdR, Herpid®, Stoxil®,
Idoxene®, Virudox® -
figure 14
This is similar to BVDU and is now used mainly in eye drops or a
topical cream for HSV keratitis.
Trifluorothymidine (Trifluridine)
Chemical name: 5-trifluoromethyl-2′-deoxyuridine
Other names: TFT, Viroptic®.
- figure 15
This is similar in its mode of action to BVDU and IDU. It also is activated by
viral thymidine kinase. TFT is used as a topical cream or in eye drops
for HSV keratitis.
|
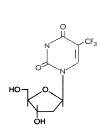 Figure 15
Figure 15
Trifluorothymidine
|
Non-nucleoside inhibitors of reverse transcriptase
(See figure 16)
Because of the problems with AZT and the other nucleoside analogs in the treatment of
HIV, interest has grown in another approach to inhibiting the same enzyme,
reverse transcriptase. Alternative drugs might be useful in combination therapy since
there is a limit to the number of mutations that reverse transcriptase can bear without
losing function. Clearly, mutations resistant to a non-nucleoside non-competitive
inhibitor of reverse transcriptase would be at a different site in the enzyme from the
mutation that makes the enzyme resistant to a competitive nucleoside analog.
Non-nucleoside inhibitors are the most potent and selective reverse transcriptase inhibitors that we have, working at nanomolar
concentrations. They have minimal toxicity in tests with cultured cells
(anti-viral activity at 10,000 to 100,000-fold lower concentration than
cytotoxic concentration) and have been shown to work synergistically with
nucleoside analogs such as AZT. Moreover, they work against nucleoside-analog
resistant HIV. Thus, these
drugs have high therapeutic index and also show good bioavailablity so that anti-viral
concentrations are readily achievable. They are non-competitive reverse
transcriptase inhibitors that target an allosteric pocket on the reverse
transcriptase molecule.
|
 Figure 16
Figure 16
Non-competitive reverse transcriptase inhibitors
|
Not surprisingly, since these drugs target reverse
transcriptase, resistant mutants
rapidly emerge, even after only a few passages in cultured cells. In patients,
resistant mutants also arise rapidly. They are therefore of little use in
monotherapy; however, although resistant virus strains are cross resistant to other
non-nucleoside reverse transcriptase inhibitors, they are not to nucleoside analog inhibitors. There is also
some evidence that these drugs may be able to overcome resistance at the high concentrations
that seem to be achievable.
There is now a collection of such agents that are chemically distinct:
Nevirapine
Chemical name: 11-cyclopropyl-5,11-dihydro-4-methyl-6H-dipyrido[3,2-b:2′,3′-f][1,4]diazepin-6-one
Other names: NVP or BIRG-587, Viramune®
In
monotherapy, this drug causes an initial fall in the number of HIV virions but
resistance sets in and virus titers rise again to a high level. This drug has been approved
for therapy in AIDS patients.
Delavirdine
Chemical name:
1-(5-methanesulfonamido-1H-indol-2-yl-carbonyl)-4-[3-(1-methylethyl-amino)pyridinyl)piperazine
monomethane sulfonate
Other name: Rescriptor®.
This is a bis (heteroaryl) piperazine
compound.
Considerable increases are observed in CD4+ cells in combination therapy
using this drug with AZT and 3TC. There have been promising results in patients with very
low CD4+ cells that have prior treatment with AZT. In combination with AZT and
3TC, DLV may delay emergence of resistance to AZT. The drug is absorbed rapidly.
DLV is used in combination with a nucleoside analog such as AZT and the
protease inhibitors discussed below.
|
|
MOLECULAR STRUCTURE
Nevirapine
Efavirenz |
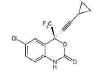 Figure 17
Figure 17
Efavirenz
(Sustiva)
|
Efavirenz
Chemical name:
(−)6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-1,4-dihydro-2H-3,1-benzoxazin-2-one
Other names: Sustiva®, Stocrin®.
Formerly known as DMP-266 - figure 17
Efavirenz used in combination with other drugs,
can suppress viral load at least as well as the protease inhibitor Indinavir in the
equivalent combination with nucleoside reverse transcriptase inhibitors. In a comparison of viral load reduction with
Efavirenz plus AZT plus
3TC, vs. a standard-of-care control group treated with Indinavir plus AZT plus
3TC, the
Efavirenz combination suppressed viral load to below 400 copies in a significantly higher
proportion of the volunteers than the control arm, at all time points between week 2 and
week 24.
|
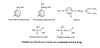
Figure 18Foscarnet
|
Other non-nucleoside polymerase inhibitors
Foscarnet
Chemical name: trisodium phosphonoformate
Other names: Foscarnet sodium, Foscavir®,
PFA, phosphono formic acid - figure 18
This is a competitive inhibitor of DNA
polymerase - it binds to pyrophosphate site. Viral DNA polymerase is
inhibited at
10-100x lower concentration than cell DNA polymerases giving some selectivity.
It is used intravenously for CMV retinitis is AIDS patients and in other
immunocompromised patients. It is useful when the infecting virus has gained
resistance to other drugs such as Acyclovir.
|
|
MOLECULAR
STRUCTURE
Foscarnet |
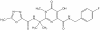 Figure 19a
Figure 19a
Isentress (Raltegravir)
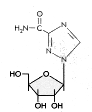 Figure 19b
Figure 19b
Ribavirin
|
DNA INTEGRATION Retroviruses copy their RNA
genome into DNA using reverse transcriptase. The DNA may remain as a circular
provirus or may be integrated into the cellular DNA. The latter is necessary for
transcription to genomic and messenger RNA . Thus, integration is required for
viral replication. Integration of viral DNA is effected by the integrase
enzyme which is encoded in the pol gene. The necessity of integration for
replication means that the integrase would be a selective drug target. Recently,
a specific integrase inhibitor has been approved.
Raltegravir
Chemical name: is
N-[(4-Fluorophenyl)methyl]-1,6-dihydro-5-hydroxy-1-methyl-2-[1-methyl-1-[[(5-methyl-1,3,4-oxadiazol-2-yl)carbonyl]amino]ethyl]-6-oxo-4-pyrimidinecarboxamide
monopotassium salt.
Other names: Isentress® ,
MK-0518 - figure 19a
Isentress can be used as part of a HAART regimen when the patient is
resistant to other drugs such as protease inhibitors. It was comparable to
Sustiva (standard of care) in HAART over a period of 24 weeks. More than 80
percent of those who took the drug showed a drop in the blood level of virus to
barely detectable levels It is not approved for HIV-infected children.
RNA SYNTHESIS INHIBITORS
Ribavirin
Chemical name: 1-β-D-ribofuranosyl-1H-1,2,4-triazole-3-carboxamide
Other names: Virazole®, Virazid®, Viramid®
- figure 19b
This drug is not a pyrimidine or
a purine. It inhibits influenza
RNA polymerase non-competitively in vitro but poorly in vivo. It may act
as a guanosine analog and inhibit 5' cap formation on mRNA. The cap normally contains
methyl guanosine. However, ribavirin is known to inhibit the production of
infectious polio virus and this virus does not have a methyl guanosine cap; so
there must be alternative mechanisms for ribavirin action. It is likely that
this drug introduces multiple mutations into viral RNA rendering it incapable of
a new round of cell infection
An aerosol form is used against RSV (respiratory syncytial virus) and
the drug is used intra-venously against Lassa fever. N.B. Ribavirin can antagonize
the effect of AZT as was found in some initial combination therapy trials against HIV.
Neplanocin A
Chemical name: 4-Cyclopentene-1,2-diol, 3-(6-amino-9H-purin-9-yl)-5-(hydroxymethyl)-,
(1S,2R,3R)-
Other names: dihydropropyl adenine, Vidarabine
This drug, a potent inhibitor of S-adenosylhomocysteine hydrolase, may also inhibit capping of mRNA.
S-adenosylhomocysteine hydrolase inhibitors have been shown to exert
anti-viral activity against pox-, paramyxo-, rhabdo-, filo-, bunya-, arena-, and
reoviruses. They also interfere with the replication of HIV by inhibition of the
Tat transactivation process.
Sofosbuvir
This drug (Gilead Sciences) inhibits
hepatitis C virus’s RNA polymerase enzyme. It
is a chain-terminating nucleotide analog which is incorporated into newly
synthesized viral RNA. Its effectiveness varies according to which genotype of
hepatitis C, infects the patient. About a quarter of patients in the United
States are infected with hepatitis C genotypes 2 and 3. These patients are
treated with sofosbuvir in combination with ribavirin but without interferon.
Since interferon has to be injected, this will be the first completely oral
treatment for hepatitis C.
More than 70% of patients infected with hepatitis C in the United States
are infected with genotype 1. They require interferon plus ribavirin
together with sofosbuvir over a period of 12 weeks. In a clinical trial,
more than 90 percent of previously untreated patients taking sofosbuvir in
combination with interferon and ribavirin showed no detectable virus in
the blood at the end of treatment.
There are a large number of other drugs that are very effective at inhibiting
Hepatitis C. These are listed on this page
|
|
MOLECULAR
STRUCTURE
Ribavirin |
|
|
RNA CLEAVAGE ENZYMES
Ribozymes are RNA molecules that have catalytic properties among which are
the specific cleavage of nucleic acids. Heptazyme is a ribozyme that cleaves
hepatitis C RNA at highly conserved regions (thereby reducing the possibility
of the development of resistance). It recognizes and cuts all known types of the
hepatitis C virus, thereby stopping viral replication. Heptazyme has not
been successful in clinical trials.
|
| |
PROTEIN SYNTHESIS INHIBITORS
Little progress has been made in the development of drugs that inhibit viral
protein synthesis since viruses use host cell translation mechanisms. However,
one drug in this class is available.
Fomivirsen
Chemical name: Anti-sense oligonucleotide
Other names: ISIS 2922, Vitravene®.
Fomivirsin is an anti-sense oligonucleotide made of 21 nucleosides that are
phosphorothioate stabilized. It can be administered as an intra-ocular injection
for CMV retinitis. It specifically hybridizes to the mRNA for CMV immediate
early 2 protein, blocking its translation.
|
 Figure 20
Figure 20
The process of retrovirus protease activity in which the protease starts
as part of the POL polyprotein and then cleaves the polyprotein
|
PROTEIN PROCESSING INHIBITORS
Protease inhibitors
Many viruses must cleave the proteins that they make. In the case of surface
glycoproteins, this is usually carried out by a host protease in the secretory pathway
(e.g. in Golgi body). In the case of internal proteins, such as the polymerase or the
group-specific antigens (GAGs) of retroviruses and some other viruses, there is a viral protease that is encoded in the POL gene (figure 20).
Active site-directed inhibitors of the HIV aspartyl protease have been developed as
this enzyme is not similar to known host proteolytic enzymes and therefore the
inhibitors should show specificity to viral proteins. The action of the HIV
protease is crucial to viral infectivity. Now we have the promise of a drug regimen that can suppress indefinitely the progress
of disease.(see also
anti-HIV drugs)
The anti-HIV protease inhibitors are all substrate analogs (figure 22). When used individually they can drive down viral load
to between one 30th and one 100th of initial value but
sub-optimal doses of these inhibitors, when used alone, can result in loss of suppression
after several months and an accumulation of multiple mutations in the protease gene giving
a high level of drug resistance. However, it should be noted that patients with sustained suppression do not
develop the resistant mutations. This seems to be because replication must be maintained
for the development of such mutations under the selective pressure of the drug.
|
|
|
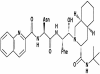 Figure 21
Figure 21
Saquinavir
|
Saquinavir
(SQ)
Chemical name: cis-N-tert-butyl-decahydro-2-[2(R)-hydroxy-4-phenyl-3(S)-[[N-2-quinolylcarbonyl-L-asparaginyl]-amino]butyl]-(4aS–8aS)-isoquinoline-3(S)-carboxamide
methane sulfonate
Other names: Invirase® (hard gel capsules), Fortovase®
soft gelatin capsules.
(Hoffman-La Roche,
figure 21).
This is a hydroxyethylamine transition-state
analog of the cleavage site on a protein recognized by the HIV protease. It is the least
bio-available of the present protease inhibitors and is the least effective. Nevertheless,
SQ + AZT + ddC reduced viremia with a rise in T4 cells in individuals with a T4 cell count
of 50 - 300/mm3.
Ritonavir
Chemical name:
[5S-(5R,8R,10R,11R)]-10-hydroxy-2-methyl-5-(1-methylethyl)-1-[2-(methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12-tetraazatridecan-13-oic
acid 5-thiazolylmethyl ester
Other names: Norvir®
(Abbot Labs).
This drug reduces AIDS-defining events and death by 58% compared to
placebo. It causes nausea in 25% of patients. It is used as part of a triple
drug highly active anti-retroviral therapy (HAART).
Indinavir
Chemical name:
[(1S,2R,5(S)-2,3,5-trideoxy-N-(2,3-dihydro-2-hydroxy-1H-inden-1-yl)-5-[2-[[(1,1-dimethylethyl)amino]carbonyl]-4-pyridinylmethyl)-1-piperazinyl]-2-(phenylmethyl- -erythro)pentonamide -erythro)pentonamide
Other names: Crixivan®. (Merke).
Indinavir plus two anti-RT drugs (HAART) reduces HIV to such an
extent that PCR cannot detect the virus in 85% of patients
Amprenavir
Chemical name: 3S)-tetrahydro-3-furyl-N-[(S,2R)-3-(4-amino-N-isobutylbenzene-sulfonamido)-1-benzyl-2-hydroxypropyl]carbamate
Other names: , Agenerase®, Prozei® (Glaxo)
This is another
protease inhibitor used in combination HAART therapy
Nelfinavir
Chemical name: [3S-(3R,4aR,8aR,2′S)]-2-[2′-hydroxy-3′-phenylthiomethyl-4′-aza-5′-oxo-5′-[2′-methyl-3′-hydroxyphenyl)-pentyl]-3-(N-(tert-butyl)-carboxamide)-decahydro
isoquinoline methane sulfonate
Other names: Viracept®.
Lopinavir
Chemical name: N-(4(S)-(2-(2,6-dimethylphenoxy)-acetylamino)-3(S)-hydroxy-5-phenyl-1(S)-benzylpentyl)-3-methyl-2(S)-(2-oxo(1,3-diazaperhydroinyl)butanamine
Other names: ABT-378/r, Kaletra®.
Lopinavir is administered combined with Ritonavir, another protease
inhibitor at a 4/1 ratio. Again, it is used as part of HAART.
Atazanavir
Chemical name: 1-[4-(pyridin-2-yl)phenyl]-5(S)-2,5-bis-{[N-(methoxycarbonyl)- -tert-leucinyl]amino}-4(S)-hydroxy-6-phenyl-2-azahexane -tert-leucinyl]amino}-4(S)-hydroxy-6-phenyl-2-azahexane
Other names: CGP 73547, BMS-232632, Reyataz®.
(Bristol-Myers Squibb)
Bevirimat
Chemical name: 3-O-(3′,3′-dimethylsuccinyl) betulinic
acid
Other names: PA-457 (Panacos Pharmaceuticals)
The protease inhibitors described above are general inhibitors of the HIV
aspartyl protease. Bevirimat is more specific but is also involved in the
maturation of the virus.
The assembly of the HIV virus budded from the cell into an infectious virion
depends upon Pr55Gag, a precursor of the Gag proteins. Pr55Gag is assembled
into the virus particle which buds from the cell and at the same time a
maturation process occurs in which the viral protease cleaves P55Gag to
generate several smaller proteins including the immature capsid protein, the
matrix protein, the nucleocapsid protein and p6. The immature capsid protein
(p25) is cleaved to form mature capsid protein (p24). This maturation
process results from a structural rearrangement in which the electron-dense
conical core of the mature virion is formed. Bevirimat inhibits the cleavage
that occurs in the maturation of p25 to p24. Specifically, the cleavage of
the p25 to p24 is disrupted, resulting in the formation of defective,
noninfectious virus particles.
|
|
MOLECULAR
STRUCTURE
Indinavir |
Highly active anti-retroviral therapies
(HAART)
Combination therapies (triple drug cocktail, HAART)
are very effective and can reduce viral load in the patient below detectable
levels implying that HIV replication has ceased. One such HAART cocktail consists of zidovudine (AZT) ,
lamivudine (3TC), both nucleoside analog reverse transcriptase inhibitors, and
Indinavir, a protease inhibitor. Viral RNA levels before treatment, which may be
as high as 11 million copies per ml, are reduced to undetectable levels in few
weeks by this drug combination (we can measure as low as 20 copies /ml) (figure
23). The evidence suggests that there is NO
replicating virus in these patients and this is sustained for several years.
When treatment is stopped, however, the virus comes back because of latent virus
in memory T cells and possibly other cells.
Another triple drug combination consists of two
nucleoside analog reverse transcriptase inhibitors (tenofovir,
(R)-9-(2-Phosphonylmethoxypropyl)adenine) and emtricitabine
(2',3'-Dideoxy-5-fluoro-3'-thiacytidine) plus the non-nucleoside inhibitors of
reverse transcriptase, efavirenz (Sustiva).
The trouble with all of these complicated drug regimens is compliance.
The
components of HAART must be taken at different times, sometimes in the
middle of the night as well as during the day sand must be taken with
different foods. For example, failure to take saquinavir within 2 hours of high fat meal leads to no absorption of drug. On the other
hand, Indinavir must be ingested with minimal food intake.
In patients that fail to take the three drugs for a week, there is a marked rise in
viral load. Non-compliance with protease inhibitor therapy is of serious concern
as the new virus that
emerges is resistant to the inhibitor being taken and also resistant to other
protease inhibitors. This is a major problem since the new resistant mutants may be
transmitted to others. Thus if a patient is known to be likely to be non-compliant he/she
should probably not be offered the drugs since resistance can emerge so quickly and can be
spread to contacts.
|
|
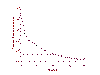 Figure 22
Figure 22
Level of HIV RNA in serum as measured by PCR after treatment with HAART
|
 Figure 23
Figure 23
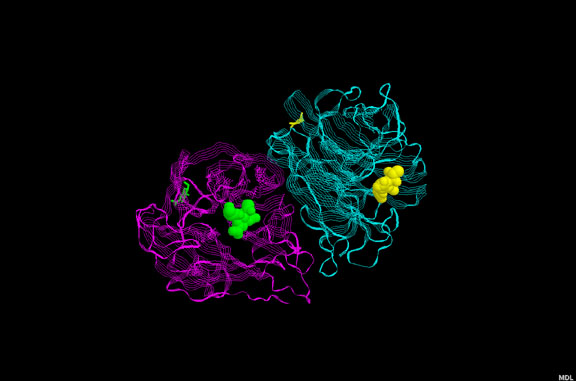
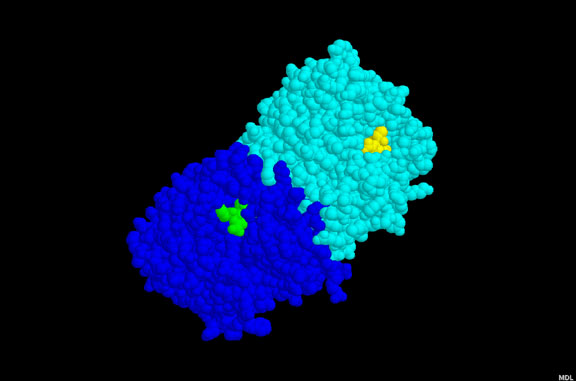
This diagram derived from X-ray crystallography shows the dimeric HIV aspartyl protease
(ribbons). Aspartate residues are shown as ball and sticks. Note that four aspartates are
clustered at the active site of the enzyme. A protease inhibitor is shown fitting into the
active site
|
Can we cure an HIV infection with drug therapy?
Some
years ago this possibility would have
been scoffed at. The drugs available then reduced viral load only to small extent and a
double drug combination was thought to be acting well if it led to a rise in CD4 cells of
50/cu mm and the viral load was down 1.5 logs. Now these are considered to be
very small changes. If, as seems likely, the triple drug therapy when taken
correctly stops all HIV replication in the patient, we might be able to
eliminate the virus as cells that harbor it in the latent form are turned over.
There is evidence,
however, that this may be difficult because latent
reservoirs of HIV undoubtedly exist. When a CD4 cell leaves the thymus it
may
meet an antigen, activate and subsequently die but a small subset of these cells become memory T cells and
revert to a resting state. They may stay in the body for many years and if they
are HIV-infected they will harbor the provirus. These cells therefore form a reservoir for HIV in the
patient. The infection
rate of this subset of cells does not appear to be great, less than 1 in 10,000 harbor
latent viral DNA. This means that only some 10,000,000 of the 1000 trillion lymphocytes in
the body are latently infected. But these may persist of decades and they
will be
untouched by the triple therapy combination. In individuals that have been treated with the
combination therapy for more than 3 years, the rate of latently infected cells remains the same (1
in 10,000). Interestingly, the archival virus had the same resistance
patterns as those that infected the patient. This means that in more than 3 years there were
probably no new rounds of HIV replication. However, the bad news is that this
reservoir of cells may
last decades.
|


 Figure 24
The requirement for neuraminidase in the life cycle of influenza virus
Figure 24
The requirement for neuraminidase in the life cycle of influenza virus
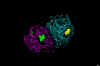
 Figure
25
Figure
25
Influenza virus neuraminidase complexed with Relenza.
Left: The enzyme is shown as strands. Relenza is space-filled. The single
N-acetyl glucosamine residue on each chain of the dimer is shown as ball
and stick. Right: The enzyme is space-filled showing the inhibitor at
the active site in a cleft in the surface of the molecule
|
PROTEIN MODIFICATION INHIBITORS
- Glycosylation
2-deoxyglucose and D-glucosamine interfere with glycosylation
in
vitro but, not surprisingly, have little effect in vivo. Castanospermine
(a natural product derived from a species of Australian chestnut) interferes with glycosylation of HIV and other retroviruses. It
leads to a dramatic decrease in syncytia. Interest in this drug as an anti-HIV agent has
waned.
- Phosphorylation
No good drugs that target
viruses by altering the phosphorylation of their proteins
have been found
- Sialidation
Two glycoproteins are found on the surface of
influenza viruses; the hemagglutinin and the neuraminidase (sialidase). The latter has
several functions. It allows the virus to move through mucous secretions in the
respiratory tract so that it may infect new cells. Since sialic acid is the influenza
receptor, it is necessary to remove sialic acid from the surface of the infected cell and
of the virus so that viral particles may escape (figure 24). The neuraminidase is therefore very
important for the spread of the virus from cell to cell.
Zanamivir
Chemical name:
4-guanidino-2,4-dideoxy-2,3-didehydro-N-acetylneuraminic
acid,
5-acetylamino-4-[(aminoiminomethyl)amino]-2,6-anhydro-3,4,5-trideoxy-D-glycero-D-galacto-non-2-enonic
acid
Other names: CG 167, Relenza®
Zanamivir is an anti-viral agent for influenza announced in the fall of 1997.
It is a
potent inhibitor of the viral neuraminidase of types A and B influenza viruses
(figure 25).
This
is important as the previously available drugs such as rimantadine are ineffective against
influenza type B. The design of Zanamivir is based on the three-dimensional structure of
the neuraminidase. Treatment of community-acquired type A and B influenza with
Zanamivir shortens the duration of major symptoms by about one day in the study group as a whole
and about three days in sicker patients if the drug is started early.
Since no antiviral drug has been approved for the treatment or prevention of influenza
B, Zanamivir could fill a niche in the control of influenza, but type B causes only about
35 percent of cases. Moreover, it has the disadvantage of requiring aerosol delivery to
the respiratory tract, an approach that could prove difficult for many.
Oseltamivir
Chemical name:
ethyl ester of (3R,4R,5S)-4-acetamido-5-amino-3-(1-ethylpropoxy)-1-cyclohexane-1-carboxylic
acid
Other names: GS 4104, Ro 64-0796, Tamiflu®
Another neuraminidase
inhibitor, Oseltamivir is a carbocyclic sialic acid analogue that can be given orally.
OTHER
TARGETS In the retrovirus life cycle, the targeting of the specific
protease that is necessary for the formation of an infectious virus particle has
been particularly successful. Earlier, reverse transcriptase inhibitors had also
been successful but the nucleoside analogs cause severe side effects because
they also inhibit the host's DNA polymerase. In contrast, the non-nucleoside
inhibitors of reverse transcriptase show excellent therapeutic indices. In each
case, however, monotherapy leads to the rapid emergence of resistant mutants. Many other
possible targets for intervention in the life cycles of viruses are under
investigation and, of course, the goal is specificity. In the case of the
retroviruses, in addition to those drugs described above, inhibitors of the integrase
are being extensively studied but none has yet made it to the clinic
as routine treatment. |
|
|
|
MOLECULAR
STRUCTURE
Castanospermine
Zanamivir Relenza
Oseltamivir |
|
|
|
|
 Return to the
Virology section of Microbiology and Immunology On-line Return to the
Virology section of Microbiology and Immunology On-line
Page maintained by
Richard Hunt
|
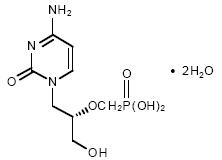
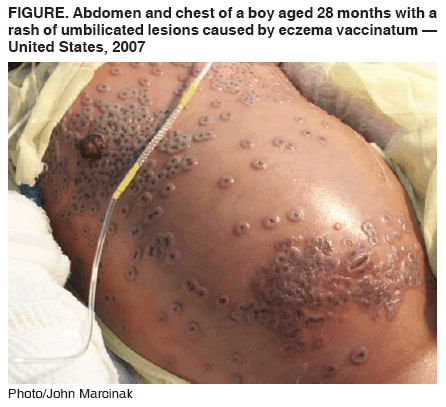
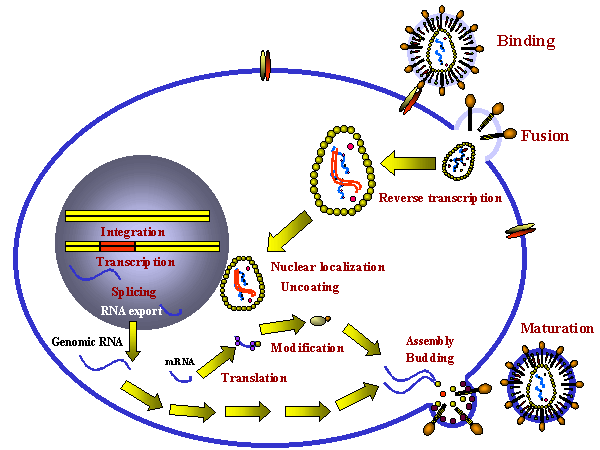 Figure 1
Figure 1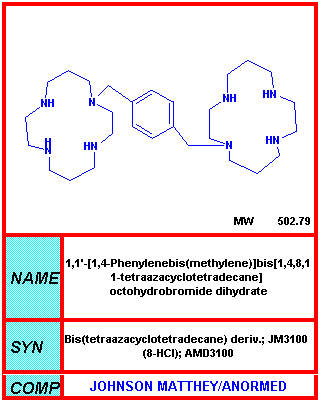 Figure 1a
Figure 1a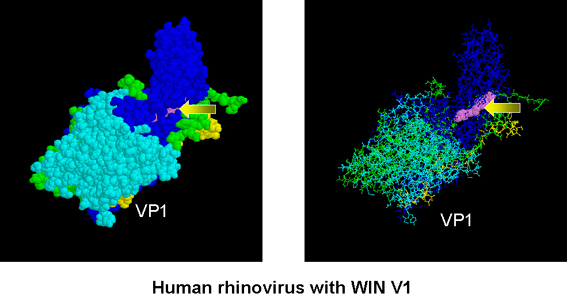 Figure 3
Figure 3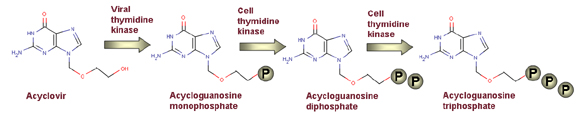 Figure 5
Figure 5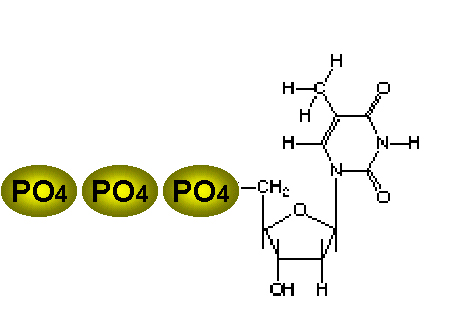 Figure 6
Figure 6 Figure 7
Figure 7 Figure 8
Figure 8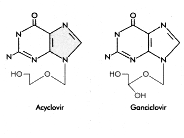 Figure 9
Figure 9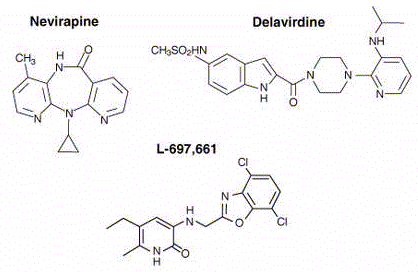 Figure 16
Figure 16 Figure 17
Figure 17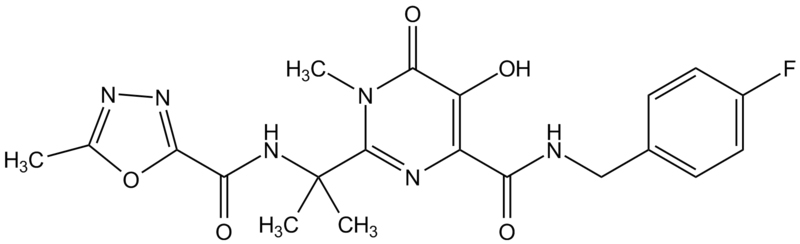 Figure 19a
Figure 19a Figure 20
Figure 20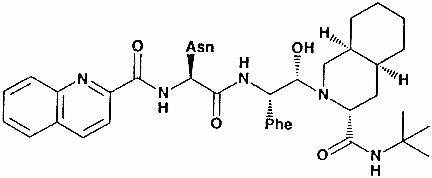 Figure 21
Figure 21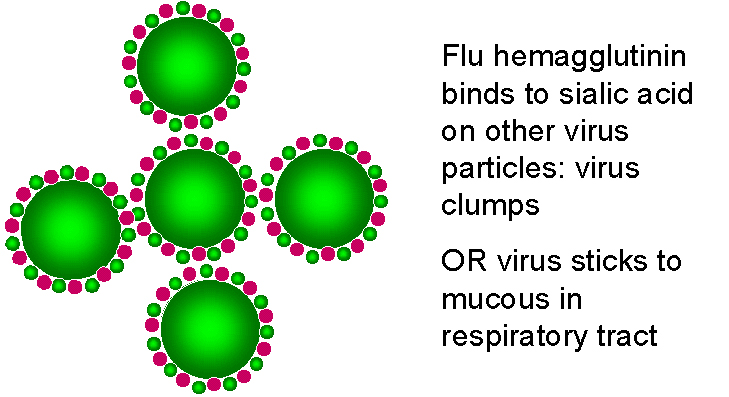
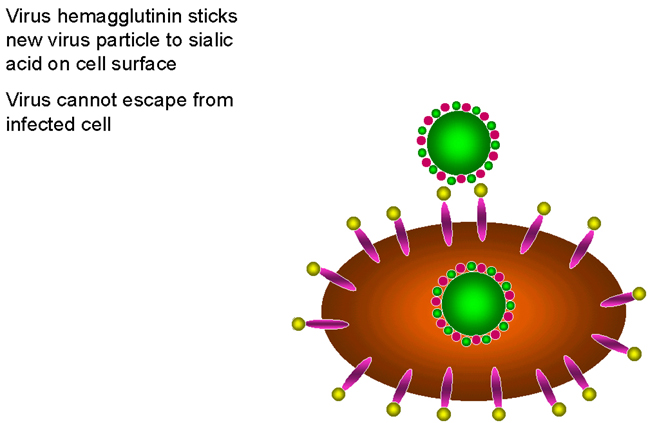
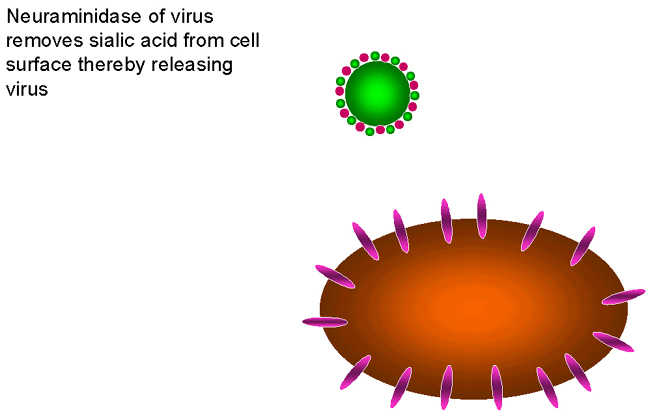 Figure 24
The requirement for neuraminidase in the life cycle of influenza virus
Figure 24
The requirement for neuraminidase in the life cycle of influenza virus