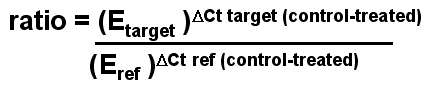| x | x | ||||||||||
|
|
 |
||||||||||
| x | x | ||||||||||
|
|
To see larger images, click on the image which will be enlarged in a pop-up window. You must close this window before opening another. This does not yet apply to the first twelve images. These appear on a new page | ||||||||||
|
FEEDBACK |
|
||||||||||
|
Polymerase chain reaction (PCR) is a method that allows exponential
amplification of short DNA sequences (usually 100 to 600 bases) within a
longer double stranded DNA molecule. PCR entails the use of a pair of
primers, each about 20 nucleotides in length, that are complementary to
a defined sequence on each of the two strands of the DNA. These primers
are extended by a DNA polymerase so that a copy is made of the
designated sequence. After making this copy, the same primers can be
used again, not only to make another copy of the input DNA strand but
also of the short copy made in the first round of synthesis. This leads
to exponential amplification. Since it is necessary to raise the
temperature to separate the two strands of the double strand DNA in each
round of the amplification process, a major step forward was the
discovery of a thermo-stable DNA polymerase (Taq polymerase) that was
isolated from Thermus aquaticus, a bacterium that grows in hot
pools; as a result it is not necessary to add new polymerase in every
round of amplification. After several (often about 40) rounds of
amplification, the PCR product is analyzed on an agarose gel and is
abundant enough to be detected with an ethidium bromide stain. For
reasons that will be outlined below, this method of analysis is at best
semi-quantitative and, in many cases, the amount of product is not
related to the amount of input DNA making this type of PCR a qualitative
tool for detecting the presence or absence of a particular DNA. In order
to measure messenger RNA (mRNA), the method was extended using reverse
transcriptase to convert mRNA into complementary DNA (cDNA) which was
then amplified by PCR and, again analyzed by agarose gel
electrophoresis. In many cases this method has been used to measure the
levels of a particular mRNA under different conditions but the method is
actually even less quantitative than PCR of DNA because of the extra
reverse transcriptase step. Reverse transcriptase-PCR analysis of mRNA
is often referred to as "RT-PCR" which is unfortunate as it can be
confused with "real time-PCR". As we shall see below, one reason that makes reverse transcriptase-PCR, as usually practiced, non-quantitative is that ethidium bromide is a rather insensitive stain. Methods such as competitive PCR were developed to make the method more quantitative but they are very cumbersome and time-consuming to perform. Thus, real-time PCR (or reverse transcriptase real-time PCR) was developed.
|
|||||||||||
|
USEFUL LINKS |
|||||||||||
 |
|||||||||||
| Abstract of the original PCR paper | |||||||||||
| First, let us review reverse transcriptase PCR in more detail | |||||||||||
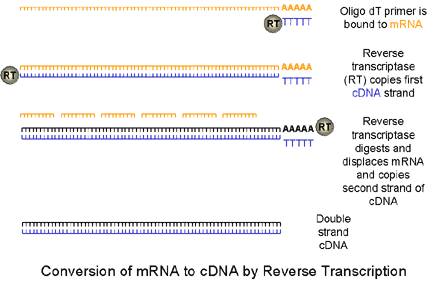
|
Polymerase
chain reaction (PCR) allows the exponential copying of part of a
DNA molecule using a DNA polymerase enzyme that is tolerant to
elevated temperatures.
1. mRNA is copied to cDNA by reverse transcriptase using an oligo dT primer (random oligomers may also be used). In real-time PCR, we usually use a reverse transcriptase that has an endo H activity. This removes the mRNA allowing the second strand of DNA to be formed. A PCR mix is then set up which includes a heat-stable polymerase (such as Taq polymerase), specific primers for the gene of interest, deoxynucleotides and a suitable buffer. |
||||||||||

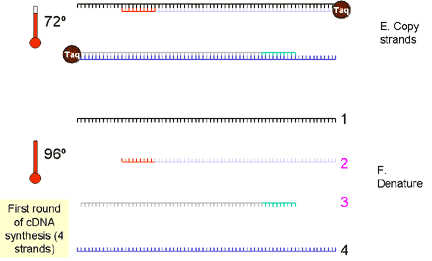
|
2.
cDNA is denatured at more than 90 degrees (~94 degrees) so that the two strands separate. The
sample is cooled to 50 to 60 degrees and specific primers are annealed
that are complementary to a site on each strand. The primers sites may be
up to 600 bases apart but are often about 100 bases apart, especially when
real-time PCR is used.
3. The temperature is raised to 72 degrees and the heat-stable Taq DNA polymerase extends the DNA from the primers. Now we have four cDNA strands (from the original two). These are denatured again at approximately 94 degrees. |
||||||||||
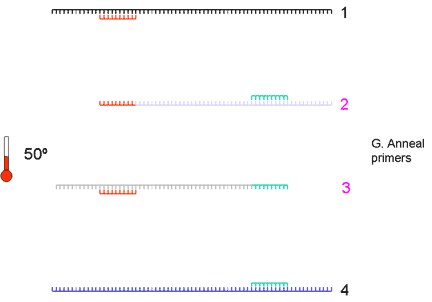
|
4. Again, the primers are annealed at a suitable temperature (somewhere between 50 and 60 degrees) | ||||||||||
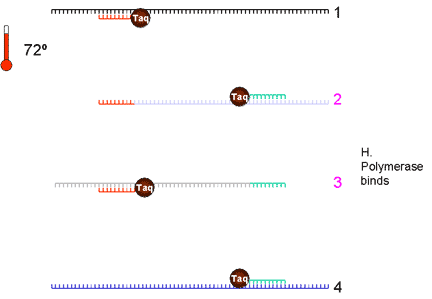

|
5. Taq DNA polymerase binds and extends from the primer to the end of the cDNA strand. There are now eight cDNA strands
|
||||||||||
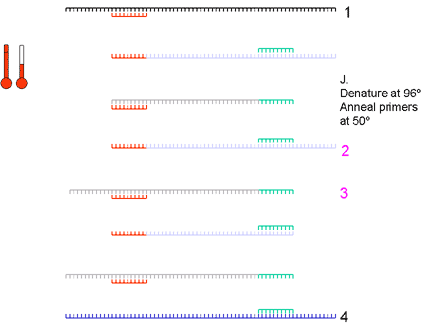
|
6. Again, the strands are denatured by raising the temperature to 94 degrees and then the primers are annealed at 60 degrees | ||||||||||

|
7. The temperature is raised and the polymerase copies the eight strands to sixteen strands | ||||||||||

|
8. The strands are denatured and primers are annealed | ||||||||||
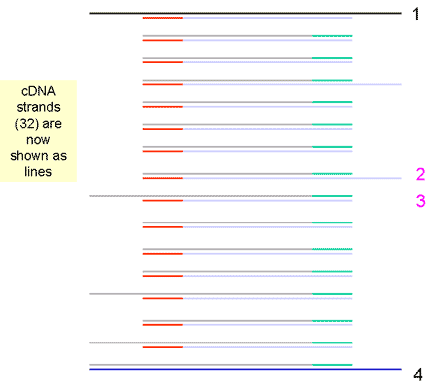
|
9. The fourth cycle results in 32 strands | ||||||||||
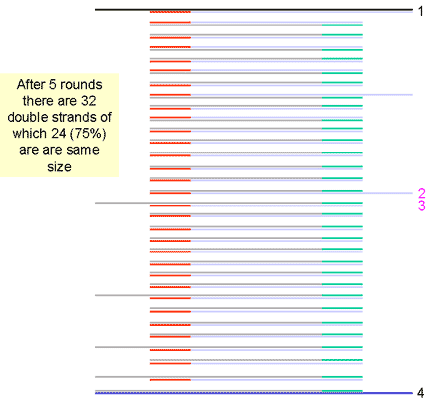
|
10. Another round doubles the number of single stands to 64. Of the 32 double stranded cDNA molecules at this stage, 75% are the same size, that is the size of the distance between the two primers. The number of cDNA molecules of this size doubles at each round of synthesis (exponentially) while the strands of larger size only increase arithmetically and are soon a small proportion of the total number of molecules. | ||||||||||
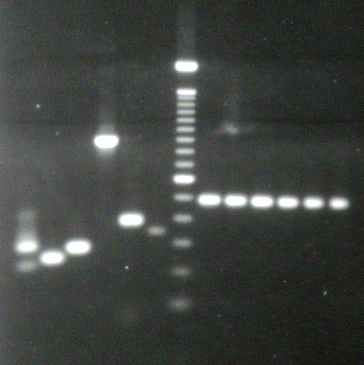 An agarose gel (1% Trevigel) stained with ethidium
bromide and illuminated with UV light which causes the intercalated stain
to fluoresce. The central lane shows markers that increase in size by
100bp. To the left are different PCR products using different primer
pairs. To the right are control reactions using primers for actin
An agarose gel (1% Trevigel) stained with ethidium
bromide and illuminated with UV light which causes the intercalated stain
to fluoresce. The central lane shows markers that increase in size by
100bp. To the left are different PCR products using different primer
pairs. To the right are control reactions using primers for actin
|
After
30 to 40 rounds of synthesis of cDNA, the reaction products are usually
analyzed by agarose gel electrophoresis. The gel is stained with ethidium
bromide
|
||||||||||
|
POWERPOINT
TUTORIAL |
This type of agarose gel-based analysis of cDNA products of reverse transcriptase-PCR does not allow accurate quantitation since ethidium bromide is rather insensitive and when a band is detectable, the exponential stage of amplification is over. This problem will be addressed in more detail below. | ||||||||||
|
Ethidium bromide is a
dye that binds to double stranded DNA by interpolation (intercalation)
between the base pairs. Here it fluoresces when irradiated in the UV part
of the spectrum. However, the fluorescence is not very bright. Other dyes
such as SYBR green, which are much more fluorescent than ethidium bromide,
are used in real time PCR. |
|||||||||||
|
SYBR Green fluoresces brightly only when bound to double stranded DNA |
In this presentation, we shall be using SYBR green to monitor DNA synthesis. SYBR green is a dye that binds to double stranded DNA but not to single-stranded DNA and is frequently used in real-time PCR reactions. When it is bound to double stranded DNA it fluoresces very brightly (much more brightly than ethidium bromide). We also use SYBR green because the ratio of fluorescence in the presence of double-stranded DNA to the fluorescence in the presence of single-stranded DNA is much higher that the ratio for ethidium bromide. Other methods can be used to detect the product during real-time PCR, but will not be discussed here. However, many of the principles discussed below apply to any real-time PCR reaction. Now let us turn to real time PCR and, first, to why it was developed |
||||||||||
|
REAL TIME PCR As we noted above, normal reverse transcriptase PCR is only semi-quantitative at best because, in part, of the insensitivity of ethidium bromide. Thus real time PCR was developed because of:
There are a variety of methods for the quantitation of mRNA. These include:• PCR is the most sensitive method and can discriminate closely related mRNAs. It is technically simple but, as mentioned above, it is difficult to get truly quantitative results using conventional PCR. |
|||||||||||
|
PROTOCOLS |
|||||||||||
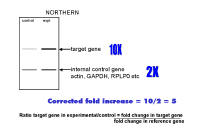 |
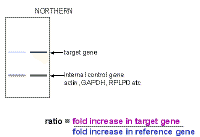
So how
do we use real-time PCR to quantitate the amount of DNA or cDNA? We can express this in a more general fashion:
|
||||||||||
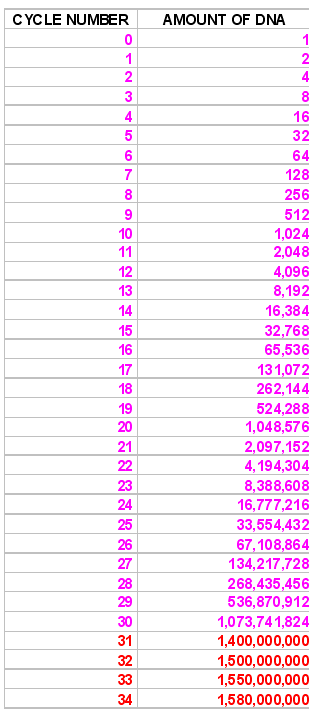
|
This brings us to the topic of standard or reference genes. A gene that is to be
used as a loading control (or internal standard) should have various
features.
However, the perfect standard does not exist; therefore whatever you decide to use as a standard or standards should be validated for your tissue - If possible, you should be able to show that it does not change significantly in expression when your cells or tissues are subjected to the experimental variables you plan to use. Commonly used standards are:
Now we need to think about the nature of the PCR reaction to understand real time QUANTITATION. The amount of DNA theoretically doubles with every cycle of PCR, as shown at the left. After each cycle , the amount of DNA is twice what it was before, so after two cycles we have 2 X 2 times as much, after 3 cycles - 2 X 2 X 2 times as much or 8 (23) times as much, after 4 cycles 2 X 2 X 2 X 2 times as much or 16 times (24) as much. Thus, after N cycles we shall have 2N times as much. But, of course, the reaction cannot go on forever, and it eventually tails off and reaches a plateau phase, as shown by the figures in red. | ||||||||||
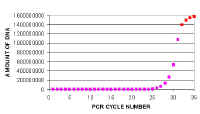
|
If we plot these figures in the standard fashion (left). We cannot detect the amplification in the earlier cycles because the changes do not show up on this scale. Eventually you see the last few cycles of the linear phase (pink) as they rise above the baseline and then the non-linear or plateau phase (red) - Actually this starts somewhat earlier than is shown here. |
||||||||||
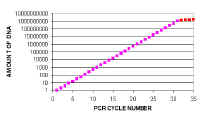 |
However, if we plot these values on a logarithmic scale, we can see the small differences at earlier cycles. In real time PCR we use both types of graph to examine the data. Note that there is a straight line relationship between the amount of DNA and cycle number when you look on a logarithmic scale. This is because PCR amplification is a exponential reaction. | ||||||||||
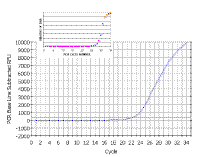 |
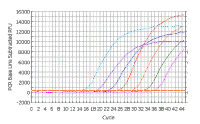
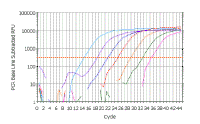
In the following discussion, the results shown will be those obtained using a Bio-Rad ICycler real-time PCR instrument; however, analysis with other machines is similar. Here is a real time PCR trace for a single well on a 96-well plate; cycle number is shown along the X-axis and arbitrary fluorescence units (actually these are fold increase over background fluorescence) are shown on the Y-axis. You can see that this mimics our theoretical graph (inset) - except that the transition to the plateau phase is more gradual. This experiment - and everything we are going to discuss - was done with SYBR Green, which has very low fluorescence in the absence of double stranded DNA and very high fluorescence in the presence of double stranded DNA. |
||||||||||
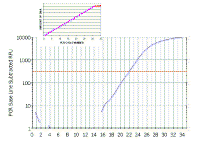 |
Here is the same real time PCR trace shown on a logarithmic scale - again it mimics our theoretical curve (inset).
|
||||||||||
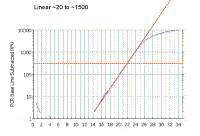 |
As we saw with the theoretical curves, you should get a straight line relationship in the linear part of the PCR reaction. In this case the reaction is linear from ~20 to ~1500 arbitrary fluorescence units. |
||||||||||
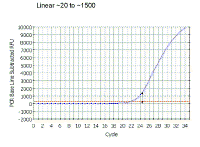 |
If we look at the same region on a regular scale, we see the linear part is, in fact, the very early part of the curve. Note that it is NOT the region which looks linear in this graphical view. This is a very important point in real-time PCR because we wish to examine the reaction while it is still in the linear phase. | ||||||||||
 |
Thus Real Time PCR is a kinetic approach in
which you look at the reaction in the early stages while it is still linear.
There are many real time machines available and the one shown at the
left is the ICycler® from BioRad. The lid slides back
to accommodate samples in a 96-well plate format. This means that we can look at a lot of samples simultaneously. The machine
contains a sensitive camera that monitors the fluorescence in each
well of the 96-well plate at frequent intervals during the PCR
Reaction. As DNA is synthesized, more SYBR Green will
bind and the fluorescence will increase. |
||||||||||
|
LINKS |
 The real-time machine is connected to a computer and software on the computer is needed to run the real time PCR machine in real-time mode |
 The plate is loaded and the lid is closed |
 Optical detection system layout of BioRad ICycler®. Image adapted from BioRad |
||||||||
|
|
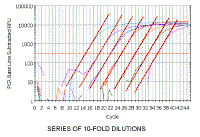
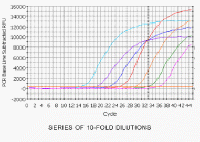
So how do we measure differences in the concentration of DNA or cDNA? This graph shows a series of 10-Fold dilutions of a sample of DNA, and as we dilute the sample, it takes more cycles before the amplification is detectable. The dark blue line here is the same sample that we have been following all along. Note that although the reactions show a series of equally-spaced curves in order of dilution as they cross the orange line, they are rather variable when we look at the upper parts of the curve. Thus, if we stopped all these reactions at, for example, 33 cycles and analyzed the products on an agarose gel, it would indicate that the blue, red and purple reactions had the same amount of amplification, even though the reactions shown by the purple and red lines differ by a factor of 100 in the amount of DNA. This emphasizes why ethidium bromide-stained gels are not quantitative and, if used to measure cDNA in PCR reactions, can give erroneous results. Thus, as we have emphasized, quantitation of the amount of cDNA in the original sample must be done where the amplification is exponential and, as we saw above, this is at the very beginning of the upturn of the curve and not in what appears to the linear region of the curve. In real time PCR, we measure the cycle number at which the increase in fluorescence (and therefore cDNA) is exponential. This is shown by the orange horizontal line in the figure (known as the threshold) and is set by the user. The point at which the fluorescence crosses the threshold is called the Ct. It should also be noted that samples that differ by a factor of 2 in the original concentration of cDNA (derived from mRNA) would be expected to be 1 cycle apart. Thus samples that differ by a factor of 10 (as in our dilution series) would be ~3.3 cycles apart. |
||||||||||
|
|
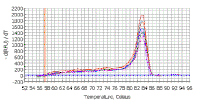
WHAT IS BEING AMPLIFIED IN OUR REACTION? The real-time machine not only monitors DNA synthesis during the PCR, it also determines the melting point of the product at the end of the amplification reactions. The melting temperature of a DNA double helix depends on its base composition (and its length if it is very short). All PCR products for a particular primer pair should have the same melting temperature - unless there is contamination, mispriming1, primer-dimer2 artifacts, or some other problem. Since SYBR green does not distinguish between one DNA and another, an important means of quality control is to check that all samples have a similar melting temperature. After real time PCR amplification, the machine can be programmed to do a melt curve, in which the temperature is raised by a fraction of a degree and the change in fluorescence is measured. At the melting point, the two strands of DNA will separate and the fluorescence rapidly decreases. The software plots the rate of change of the relative fluorescence units (RFU) with time (T) (-d(RFU)/dT) on the Y-axis versus the temperature on the X-axis, and this will peak at the melting temperature (Tm). At the left are the melting curves for the samples on the previous picture; a primer-dimer artifact would give a peak with a lower melting temperature (because it is such a short DNA). If the peaks are not similar, this might suggest contamination, mispriming, primer-dimer artifact etc. You need to be sure that the only thing you detect with SYBR green is the thing you want to detect; that is a specific DNA fragment corresponding to the size predicted from the position of the primers on the cDNA (if you are looking at mRNA) or the genomic DNA, plasmid DNA, etc (according to what your target DNA is).
|
||||||||||
|
|
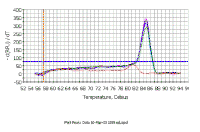
In this melting curve, all samples were run with the same primer pair, but the sample which contained no DNA (the red line) shows a melting curve with a lower Tm that the other samples; this is probably due to a primer-dimer artifact. With the SYBR green method, primer dimer artifacts are a problem since you are measuring total DNA synthesis and you need to be sure that you are measuring a Ct due to the real target for amplification. Fortunately, there are no signs of primer dimer artifacts in samples containing DNA in the graph at the left. However, this does stress the importance of primer design when using SYBR green. |
||||||||||
 |
So let's get back to the kinetics of SYBR green incorporation in our
series of 10-fold dilutions. Here are the data on an arithmetic scale. |
||||||||||
|
SERIES OF 10-FOLD DILUTIONS
|
This shows the same data in the previous picture but on a logarithmic scale.
The even spacing of the reactions is now much more obvious.
What the software measures for each sample is the cycle number at which
the fluorescence crosses the arbitrary line, the threshold, shown in orange.
This crossing point is the Ct value. More dilute samples will cross at later Ct values. |
||||||||||
|
|
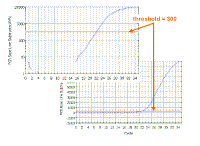
As we saw, it is important that the threshold should be in
the linear part of the reaction - this is easier to see in the
logarithmic view, where it should be no more than half way up the
linear part; in the regular view, the threshold will be close to the
bottom of the curve. However, the threshold should be high enough that
you are sure that reactions cross the line due to amplification rather
than noise. We find that if the plateau values are 4000 to to 15000, a
threshold of 300 usually works well.
We use the same threshold for all the samples in
the same experiment on the same plate. |
||||||||||
|
|
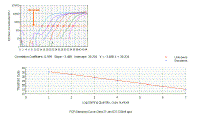
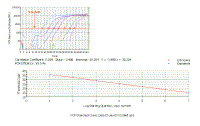
We can plot the Ct values for the dilutions against concentration - the result is a linear graph.
It should have an excellent correlation coefficient (certainly more than 0.990). |
||||||||||
| QUANTITATION OF mRNA LEVELS USING REAL TIME PCR | |||||||||||
|
|
STANDARD CURVE METHOD There are several methods to quantitate
alterations in mRNA levels using real time PCR. Let's look at the
standard curve method first. |
||||||||||
|
|
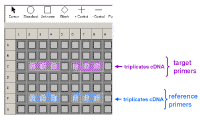
Here is an example of how we could set up a plate if we were using the standard curve method. By clicking on one of the symbols in the top line and then clicking on one of the wells in the plate diagram, we can define whether samples contain a standard (circles) or an unknown (squares) or negative control samples containing water instead of DNA (-) and whether we have single wells containing the same sample or duplicate or triplicate wells (in which case the duplicate or triplicate well are assigned the same number). The software also allows you to define your dilution factors for the standard curves, give them names, etc. The negative controls are to check that your primers and Taq polymerase/SYBR green PCR mixes are not contaminated. They also allow you to determine if your primers can form primer-dimer artifacts which are most readily seen when there is no appropriate DNA for amplification (as shown above). In this case we have extracted RNA from control
cells (C) or cells treated in some special fashion (e.g. drug treated
cells) - here designated (E) for experimental. These were then copied into cDNA using reverse transcriptase. We have set up the plate so that there
is a standard curve for the loading control (or reference gene) and also one
for the gene of interest whose expression we think may change under the
experimental conditions (the so-called target gene). We normally just do a
single point for each dilution of the standard curve since this gives a
series of points which fit very well to a straight line. We usually do the
cDNA samples in triplicate - so each sample will be done in triplicate for
each gene. |
||||||||||
|
|
You tell the software which dilution curve you want to use, and which unknowns you want it to quantitate using that curve. You do not actually give it a real copy number - just start at some arbitrary number. The machine will report the copy number or amount of DNA in each of your unknown samples and even average this for you. If you take the average of the copy number in the experimental sample, divide it by the average copy number in the control sample, this will give the fold change in the target gene. You have now calculated the upper value in the "Northern" formula we derived earlier on. Note the excellent fit of the standard curve data to a straight line - a perfect fit would have a correlation coefficient of 1.000 and here the correlation coefficient is 0.999. |
||||||||||
|
|
Similarly, you can select the wells in which you amplified the reference gene and determine the relative amounts in the experimental sample compared to the control. This will give you the bottom value in the "Northern formula" derived earlier. Now that you have both values, you
can divide the target gene value (purple) by the reference gene value
(blue) and obtain the ratio of the target gene in the experimental sample
relative to the control sample, corrected for the reference gene (loading
control). This method will therefore give the fold changes in the target and reference genes - so we can calculate a fold change corrected for any variations in the reference gene just as one would do for a northern blot. The disadvantage of this method is that you need a good dilution curve for both standard and reference genes on every plate - which would be at least 16 extra wells (including negative controls). If there is any problem with either dilution curve, the data cannot be analyzed, or a suboptimal curve has to be used. Thus, we prefer to determine efficiency accurately (on multiple days) and then take an average of multiple results and use these separately - this makes experiments simpler but you need to think a bit more about the maths of calculating the results because this time the machine does not do it for you. We find that the standard curves are highly reproducible if you use a supplier who provides a mix with stabilizer(s) for SYBR green. |
||||||||||
|
|
PFAFFL METHOD
So is there a way to do a similar calculation without doing an internal dilution curve each time? The answer is yes, we use the simple method developed by M.W. Pfaffl. However, to talk about this, we need to delve a bit more into the PCR reaction and examine the effects of the efficiency of amplification. EFFECTS OF EFFICIENCY So first of all, let's discuss the significance of efficiency |
||||||||||
|
|
|||||||||||
|
|
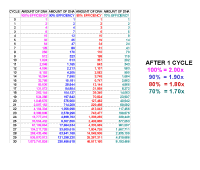
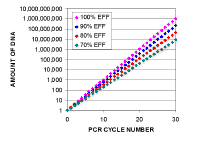
Here is a series of calculations showing how much
the DNA will be amplified if you have different efficiencies. For 100%
efficiency, there will be a doubling of the amount of DNA at each cycle, for 90% the amount
of DNA will increase from 1 to 1.9 at each cycle, so the factor is 1.9
for each cycle, and similarly for 80% and 70% it will be 1.8 and 1.7.
Notice that a small difference in efficiency makes a lot of difference in
the amount of final product. Each 10% lowering results in less than 25%
of the previous column after 30 cycles. |
||||||||||
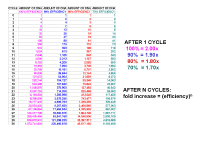
|
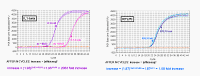
|
From this you can see that after 10 cycles the increase in DNA will be 210, if the efficiency is 100% (each cycle results in twice as much DNA), or 1.910 if each cycle results in 1.9 times as much DNA - or to generalize, after n cycles, it will be [efficiency] n. There is some ambiguity in how people define efficiency. Some people say that if you copy 90% of your DNA in a cycle, so that you end up with 1.9 times as much, the efficiency is 1.9, and this is the definition that the Pfaffl equation uses. Other people say that the efficiency is actually 0.9 since one makes 0.9 times as much. If you use this definition the fold increase will be [1 + efficiency] n. We shall use the Pfaffl definition so we won’t have to keep adding the ‘1’. |
||||||||||
|
|
This shows the effect of changes in efficiency
graphically. You can see that changes in efficiency have a major effect on
the Ct value. Note also that changes in efficiency change the slope when
you use the logarithmic scale. |
||||||||||
|
|
Since the lines diverge at higher thresholds, lower
thresholds will minimize the error due to small changes in efficiency. If a
reaction has an inhibitor of PCR in it that reduces the efficiency, the
slope will be different from unaffected reactions when you look at the
results using the logarithmic scale. Thus, if you do triplicate reactions
and one has a bad slope, you should drop that well from the analysis. |
||||||||||
|
SERIES OF 10-FOLD DILUTIONS
|
Here are the data from our dilution curve. If you are
looking at efficiencies, you want to be sure that every time you do the PCR
for the same gene you have the same slope since this is a measure of
efficiency - in this case you can see that all the samples are reasonably
close (the lines are all parallel). If there is a difference in slope of
one of your samples, it implies a problem in that tube (PCR inhibitor,
problems with the enzyme, etc). |
||||||||||
|
|
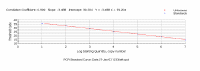
We can plot the Ct values for each of the dilutions against concentration - the result is a linear graph. It should have an excellent correlation coefficient (more than 0.990). The slope of this graph is a measure of efficiency, and can be readily used to calculate efficiency - but we shall not go into the math because the current version of the ICycler software does this for you. | ||||||||||
|
|
QUALITY
CONTROL - EFFICIENCY CURVES
Here is a list of the criteria we apply in the lab before we accept the data for efficiency from a dilution curve
If you apply the top 7 criteria, the correlation coefficient is usually not a problem - we routinely get values of 0.998 and above. |
||||||||||
|
|
We find that the 'PCR base line subtracted curve
fit' option (blue line in box at the top left), which is the default analysis mode in the current version of
the ICycler program (3.0a) does not give such good results as the 'pcr base line
subtracted' option. So we always use the latter. |
||||||||||
|
|
So how does the Pfaffl method work? Again our approach is based on the
"Northern equation". |
||||||||||
|
|
Here is the plate set up for this method. Note that this time we do not
have to include a standard curve on every plate. |
||||||||||
|
|
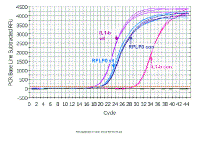
Here are the results from an experiment in which the target gene was
IL1-beta and the reference gene was RPLPO. In this experiment, we
investigated what happens when cells from the eye come in contact with
vitreous humor. RNA was extracted from control
(con) or vitreous-treated (vit) cells, and copied into cDNA. IL1-beta and
RPLP0 assays were done in triplicate on cDNAs from both control and
vitreous-treated cells. We shall start by looking at the top part of the "Northern equation" - that is determining the fold change in IL1-beta in vitreous compared to control. |
||||||||||
|
|
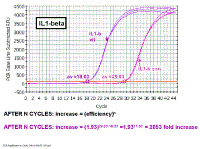
In the picture at the left, we are just looking at the results from the wells
containing the IL1-beta primers. If we look at the difference in Ct values
between the control and vitreous samples, we see there is an 11.60 cycle
difference. Earlier on we derived the equation that the change in amount of
DNA after n cycles is equal to the efficiency to the power of n. We
independently do multiple serial dilution curves on multiple days to
determine an average efficiency and we can then plug that value into the
formula. Note that when determining the difference in the Ct values
(sometimes known as the 'delta Ct'), we subtract the vitreous from the
control value - this is so that increases will have a positive result and
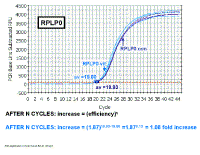
decreases a negative result for the delta Ct value. Fold increase in target gene (IL-1 beta) AFTER N CYCLES: increase = (efficiency)n AFTER N CYCLES: increase = (1.93)29.63-18.03 =1.9311.60 = 2053 fold increase We had previously shown that the efficiency of the IL-1 beta primers was 93% |
||||||||||
|
|
Here we are just looking at the RPLP0 data. Note, that, as
expected for a good reference gene, there is not much difference between
the two RNA samples with regard to their levels of RPLP0 mRNA. The same
amount of total RNA was used for reverse transcription of both RNAs, and
the same amount of each reverse transcription reaction was used for
real-time PCR. Since the Ct values are so close and the ratio of the
reference gene in the two samples is close to one, this suggests that
RPLP0 is a good reference gene for these experiments. Fold increase in 'loading control' gene (RPLPO) AFTER N CYCLES: increase = (efficiency)n AFTER N CYCLES: increase = (1.87)19.93-19.80 = (1.87)0.13 = 1.08 fold increase
|
||||||||||
|
|
So we derived the change in IL1-beta mRNA (left panel) and
in RPLP0 mRNA (right panel), and we now need to divide the change in target
gene by the change in the reference gene as we would do in the ‘Northern
blot equation’.
If we express this in a more general way as is shown below,
we get a universal formula for doing these calculations - this is the
formula in Pfaffl paper.
|
||||||||||
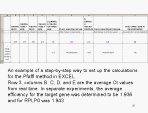 |
Here is a way to set up Excel spreadsheet to do the calculations for you.
You do not actually need all the columns, you can just write the entire
formula in one cell rather than subdividing it and spreading it across 5
cells as shown here (this was done so as to be clearer how the spreadsheet
corresponds to the calculations we have discussed). However, this does
allow you to scan readily for whether there is much change in the reference
gene. |
||||||||||
|
Delta-Delta
CT METHOD (An approximation method)
This method was one of the first methods to be used to calculate real-time
PCR results. However, as we shall see, it is an approximation method. It
makes various assumptions, and to prove that they are valid is, in our
opinion, more time consuming that doing a few extra efficiency runs for the
Pfaffl method. |
|||||||||||
|
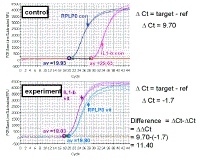
|
Let us look at the same data that we discussed before but,
for the time being, we shall ignore the data from the standard ‘loading
control’ gene. The difference between the control and treated samples for
interleukin 1-beta is shown by the red line. If we know the efficiency for
IL-1 beta
and the cycle number, we could calculate the fold change in IL-1 beta - but
there would be no loading control.
|
||||||||||
|
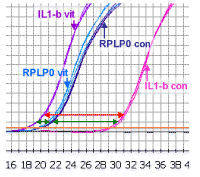
|
An approximate correction can be made for the loading control by calculating the difference between the IL1-beta Ct values and the RPLP0 values for the control samples, and then for the vitreous-treated samples (represented by the two green arrows in the picture). This makes an allowance for the fact that in the above case, there is slightly more mRNA in the vitreous-treated samples (since the RPLP0 comes up slightly earlier). This difference (or delta Ct value) is shown by the two green arrows. The difference between the two delta values represents the shift as will be seen in the next picture.
|
||||||||||
|
|
The difference between the two delta
Ct values (delta delta Ct), represents the corrected shift of the IL1-beta.
This is because, in this example, in the experimental sample the IL1-beta has moved
to the left of the standard, it has a negative value, but in math
subtraction of a negative value is equivalent to adding that value - which
makes obvious sense if you look at the diagram. The total shift
is equal to the two green arrows added together. If the experimental (vit) IL1-beta had shifted but
remained to the right of the reference curve, the value would then be
subtracted from the large green arrow to determine the shift.
|
||||||||||
|
|
Note how much different the results are when the correct efficiency is used rather than assuming 100%. The results are much closer to the ones calculated with the accurate method. However, note that the efficiency of the RPLP0 was never taken into account with this method. This method assumes that the efficiency of RPLP0 is so close to that of IL1-beta that it does not make much difference - or that the reference gene values are so similar for different RNA samples that the correction is so small that it would not be significant (it was only 1.08 fold when we calculated the change in RPLP0 cDNA for these same samples when discussing the Pfaffl method). |
||||||||||
|
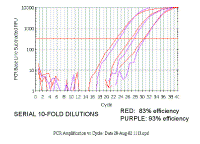
|
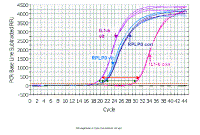
The problem with the delta delta ct is that it assumes that if you dilute the sample, the difference between the target gene and the reference gene will remain the same, so that the delta ct will be constant no matter how much you dilute the sample. However, as you can see in this figure, when we do a series of 10-fold dilutions the delta ct (difference between red and purple lines) does not remain constant - even with only 10% difference in efficiency. The purple is more efficient and gradually catches up.
|
||||||||||
|
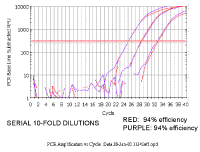
|
However, if both primer sets have a
similar efficiency, when we do a series of 10-fold dilutions the delta ct
(difference between red and purple lines) remains constant (the slight
variation for the last point here is probably because these samples were
done as single wells per sample, if replicates had been included, the
variation would be less).
|
||||||||||
|
|
SUMMARY OF EFFICIENCY(DDCt) METHOD
-- –minimal correction for the standard gene, or – that standard and target have similar efficiencies. * The 2 delta-delta Ct variant assumes efficiencies are both 100%
|
||||||||||
|
|
Special thanks to
|
||||||||||
|
Visitors:
|
| ||||||||||