|
x |
x |
|
 |
 |
|
INFECTIOUS
DISEASE |
BACTERIOLOGIE |
IMMUNOLOGIE |
MYCOLOGY |
PARASITOLOGY |
VIROLOGIE |
|
VIDEO
LECTURE
|
Virologie - Chapitre 6
Les Virus Transformants
Dr Richard Hunt
University of South Carolina School of Medicine
Columbia SC
USA
Dr Dorian McIlroy
Université de Nantes
France
|
|
|
|
PDF via PDF MYURL |
|
En
Español |
|
NË SHQIPTARE |
|
|
|
Let us know what you think
FEEDBACK |
|
SEARCH |
|

 |
|
|
|
|
|
|
Introduction
La transformation maligne (souvent raccourci en "transformation" tout
court) est le processus par lequel une cellule normale de l'organisme
devient une cellule cancéreuse. Elle peut être induite par différents
événements (par exemple, par l'exposition aux carcinogènes chimiques ou
l'irradiation ionisante), y compris par l'infection virale. La
transformation comprend souvent une perte de contrôle de la croissance,
la croissance indépendante de l'ancrage de la cellule, la capacité
d'envahir la matrice extracellulaire, la dé-différenciation et
l'immortalisation.
La perturbation des contraintes normales sur
la prolifération cellulaire qui caractérise la transformation maligne ne
peut se produire que d'un nombre de façons strictement limité, et il
peut y avoir aussi peu que quarante gènes cellulaires chez lesquels une
mutation ou une autre perturbation de leur expression conduit à une
croissance cellulaire effrénée.
Il y a deux classes de ces gènes dont
l'expression altérée peut entraîner une transformation maligne:
(a) Les gènes qui stimulent la
croissance et qui provoquent le cancer lorsqu'ils sont hyperactifs.
Des mutations dans ces gènes seront dominantes. Ces gènes sont
appelés des oncogènes.
(b) Les gènes qui inhibent la
croissance des cellules, et qui provoquent le cancer lorsqu'ils sont
inactivés. Des mutations dans ces gènes seront récessives. Ce sont
les anti-oncogènes ou des gènes suppresseurs de tumeurs.
L'étude des virus qui provoquent des cancers chez les rongeurs et chez
les oiseaux a été extrêmement importante dans l'identification des
oncogènes et des anti-oncogènes. Ce chapitre est donc divisé en deux
sections. Dans un premier temps, les principales observations chez les
virus transformants dans des systèmes modèles seront exposées, afin
d'expliquer les mécanismes par lesquels les virus oncogènes à ARN et à
ADN provoquent des cancers. Dans un deuxième temps, les virus qui sont
impliqués dans le développement de cancers humains seront présentés, et
nous verrons en quelle mesure les mécanismes observés dans des systèmes
modèles sont pertinents pour la compréhension des cancers humains
provoqués par les virus.
|
|
|
Virus transformants chez l'animal
Découverte des virus transformants
Les premiers virus impliqués dans le développement d'un cancer ont été
isolés au début du 20ème siècle par Bang et Ellerman au Danemark, et par
Peyton Rous aux Etats-Unis. Le virus isolé par Rous était capable de
provoquer la formation des sarcomes chez le poulet, d'où son nom, le "Rous
Sarcoma Virus" ou RSV. Sa découverte a été récompensée par le prix Nobel de
Médecine en 1966.
Une vingtaine d'années plus tard, les premiers virus provoquant des
cancers chez les mammifères, le virus du fibrome du lapin et le virus du
papillome de Shope, étaient découverts par Richard Shope.
Classes de virus transformants
Les premiers virus associés aux cancers ont des génomes bien distincts. Le
RSV, étant un rétrovirus, possède un génome en ARN dans la particule virale,
tandis que les virus isolés par Shope sont des virus à ADN. Cette
distinction a mené à la classification des autres virus oncogènes, qui ont
été identifiés par la suite chez les oiseaux et chez les rongeurs, en deux
groupes -
Les virus transformants à ARN, qui sont des rétrovirus, et
Les virus transformants à ADN.
Nous verrons que les stratégies de réplication de ces deux catégories de
virus sont très différentes, mais ils ont souvent un aspect de leur cycle de
réplication en commun: la capacité d'intégrer leur propre génome dans celui
de la cellule hôte. Cette intégration n'est pas toujours une étape
indispensable pour la formation de tumeurs.
|
| |
 Figure 1
Figure 1
Schéma du cycle de réplication des rétrovirus
|
Les virus transformants à ARN et la
découverte des oncogènes viraux et cellulaires
Les virus transformants à ARN
sont des rétrovirus
A la fin des années 1960, les virus transformants à ARN posaient un
problème intéressant. Il était connu que le cancer impliquait des
modifications dans l'ADN des cellules cancéreuses. Dans ce cas, comment
l'infection par un virus à ARN peut-elle mener au développement d'une
tumeur? Car pour ce faire le virus doit, d'une façon ou d'une autre,
influencer le génome de la cellule infectée – ce qui semblait impossible
pour un virus à ARN. Cette énigme fut résolue par la découverte de la
transcriptase inverse en 1970 par Howard Temin et David Baltimore (Prix
Nobel de Médecine en 1975). Les virus oncogènes à ARN étaient donc les
premiers représentants de la classe des rétrovirus, qui possèdent un
génome en ARN à l'intérieur de la particule virale qui est transcrit en
ADN à l'intérieur de la cellule hôte (Figure 1). La copie en ADN du
génome viral s’intègre ensuite dans l'ADN génomique de la cellule
infectée, et c'est ce qui permet aux rétrovirus de modifier de façon
durable les gènes exprimés au sein de la cellule hôte. Le transfert de
l'information génétique d'une molécule d'ARN vers une molécule d'ADN est
aussi une violation de ce qui avait auparavant été proposé comme le
dogme central de la biologie moléculaire; c'est à dire que l'information
biologique passe de l'ADN, puis à l'ARN, et passe enfin aux protéines.
Classification de la famille
Retroviridae
La famille Retroviridae est divisée en deux sous-familles, les
Orthoretrovirinae et les Spumavirinae.
Les Orthoretrovirinae ont classiquement été sous-divisés en
Oncovirinae et Lentivirinae, mais le groupe "Oncovirinae" rassemble des
virus assez divers, et ne semble pas représenter un vrai clade
phylogénétique. La sous-famille Orthoretrovirinae est donc divisé en
différents genres; les Alpha-, Beta-, Gamma-, Delta- et Epsilon- virus,
et les Lentivirus.
Le RSV est un Alpharetrovirus, tandis que les virus humains, HTLV-1
et HTLV-2 sont des Deltaretrovirus.
Le HTLV-1 ("Human T-cell lymphotropic virus-1"), qui est transmis par
voies sexuelle et intraveineuse, est responsable d’un type de leucémie T
de l'adulte. Cette maladie est présente dans certaines îles japonaises,
dans les Caraïbes, en Amérique latine et en Afrique.
Le HTLV-2, qui provoque la leucémie à tricholeucocytes, est endémique
dans des régions spécifiques des Amériques, en particulier chez les
populations amérindiennes.
Les Lentivirinae provoquent des maladies caractérisées par une longue
période de latence clinique avant la déclaration des symptômes. Ces
virus ont été d'abord identifiés chez les ongulés (par exemple le virus
visna, qui infecte les moutons) mais le VIH-1 (virus d'immunodéficience
humaine -1) et le VIH-2, qui sont responsables du SIDA, ainsi qu'un
grand nombre de lentivirus chez d'autres primates appartiennent à ce
groupe.
En ce qui concerne les Spumavirus, il n'existe aucune preuve de leur
pathogénicité. Ils établissent des infections persistantes dans de
nombreuses espèces animales. Ils ont été isolés à partir des primates (y
compris les humains), des bovins, des félins, des rongeurs et des
mammifères marins. Les cellules infectées par un Spumavirus ont un
aspect mousseux (en raison de nombreuses vacuoles) et forment souvent
des syncytia de cellules multinucléées géantes. Les virus mousseux
simien ("simian foamy virus") est l'espèce de type du genre Spumavirus.
Le virus mousseux humain ("human foamy virus") est une variante du virus
spumeux simien, et il est généralement acquis après une morsure de
singe.
A retenir – les virus transformants à ARN sont des rétrovirus,
mais tous les rétrovirus ne provoquent pas des cancers!
|
 Figure 2A
Figure 2A
Virus de l'immunodéficience humaine
©
Dept. de Microbiologie, Université d'Otago, Nouvelle Zélande
 Figure 2B
Figure 2B
Structure d'un retrovirus: le VIH-1.
D'après
le Harvard AIDS Institute Library of Images, Critical Path AIDS Project,
Philadelphia.
 Figure 3
Figure 3
Structure de la protéase du virus de sarcome de Rous (RSV).
Le plug-in Chime est nécessaire pour visualiser la structure.
|
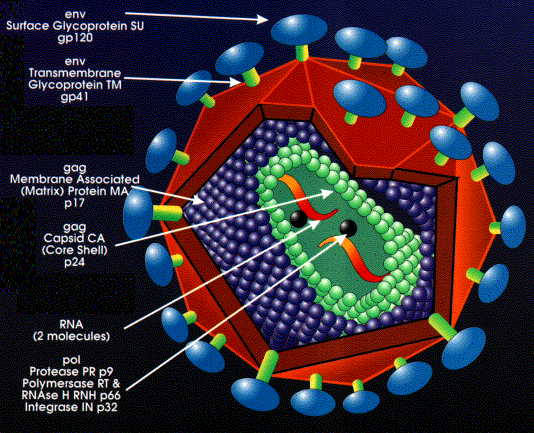
Structure des rétrovirus
Les rétrovirus sont des virus enveloppés d'un diamètre de 80 à
100 nm (Figure 2). L'enveloppe est dérivée de la membrane plasmique
de la cellule hôte, et les glycoprotéines d'enveloppe (c'est à dire,
les antigènes de surface) sont codées par gène env (enveloppe) du
virus. Le gène env code pour une protéine transmembranaire de type 1
qui est traduite par des ribosomes attachés au réticulum
endoplasmique. Par la suite, cette protéine primaire est clivée par
une enzyme de l'hôte dans l'appareil de Golgi, afin de générer deux
glycoprotéines à la surface du virus mature. Ces deux glycoprotéines
restent associées: l'une ancrée dans l'enveloppe du virus, qui
possède une structure secondaire principalement composée d'hélices
alpha et qui fonctionne comme protéine de fusion lors de l'entrée du
virus; l'autre possède une structure plus globulaire, qui fonctionne
comme glycoprotéine d'attachement.
A l'intérieur de l'enveloppe se trouve une capside icosaèdrique
composée de la protéine de capside. La protéine de matrice se trouve
à l'extérieur de la capside, tandis que l'ARN viral, associé avec la
protéine de nucléocapside, se trouve à l'intérieur de la capside.
Les protéines de matrice, de capside et de nucléocapside sont toutes
codées par le gène gag (pour "Group-specific AntiGens", ou antigènes
spécifiques de groupe) du virus. Le gène gag code pour une
polyprotéine, qui est clivée en des différentes protéines de
strucure du virus par une protéase virale codée sur le gène pol.
Chaque particule virale incorpore deux molécules d'ARN génomique,
ce qui fait que les rétrovirus sont diploïdes. L'ARN génomique est
brin (+), et comme un ARNm possède une coiffe en 5 'et une queue
polyA en 3'.
Environ 10 copies de la transcriptase inverse et de l'intégrase
sont présentes dans la particule virale mature. Ces enzymes, comme
la protéase virale, sont codées par le gène pol. Comme le gène gag,
le gène pol est d'abord traduit en une polyprotéine qui est ensuite
découpée pour libérer les différents produits matures du gène pol,
qui sont:
a) la protéase (clive les polyprotéines traduites de l'ARNm
du gène gag et du gène pol lui-même. Figure 3).
b) la transcriptase inverse (une polymérase, qui copie l'ARN
génomique du virus en ADN)
c) la RNase H (le produit initial de la transcriptase inverse
est un hétéroduplexe ARN-ADN. La RNase H clive l'ARN de cet
hétérodimère, ce qui permet à la transcriptase inverse de
synthétiser le second brin complémentaire d'ADN, et ainsi de
compléter la transcription de l'ARN simple brin du virus en une
copie en ADN double brin)
d) l'intégrase (insert le génome viral dans le génome de l'hôte)
La protéase, la transciptase inverse, et l'intégrase sont des
cibles de différentes classes de médicaments anti-rétroviraux.
|
|
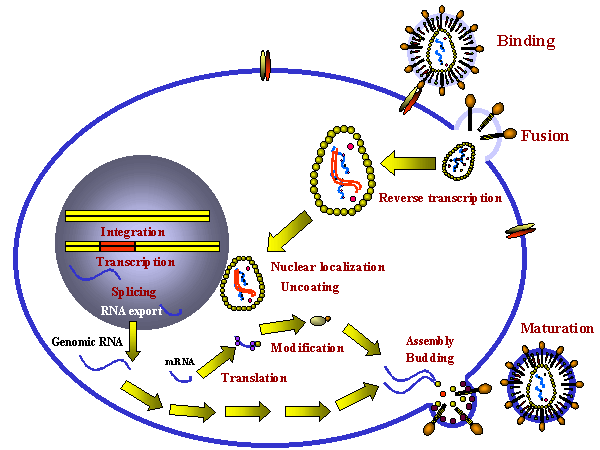
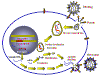 Figure 4 Etapes dans l'infection productive d'une cellule par un
rétrovirus.
Figure 4 Etapes dans l'infection productive d'une cellule par un
rétrovirus. |
Cycle de réplication des rétrovirus
Le cycle de réplication des rétrovirus (Figure 4) est composé des étapes
suivantes:
1) Attachement du virus sur son récepteur cellulaire via la
glycoprotéine d'attachement de l'enveloppe virale.
2) Entrée par endocytose ou par fusion directe avec la membrane
plasmique. L'acidification dans un endosome peut être nécessaire pour
induire la fusion, tandis que chez d'autres rétrovirus, (par exemple le
VIH) c'est l'interaction avec un co-récepteur qui permet la fusion entre
l'enveloppe virale et la membrane plasmique
3) L'ARN brin (+) est copié par la transcriptase inverse à l'ADN brin
(-). Ici, la polymérase agit comme une polymérase à ADN dépendante de
l'ARN. Puisque la transcriptase inverse est une polymérase à ADN, elle a
besoin d'une amorce. Il s'agit d'un ARNt qui est incorporé dans la
particule virale à partir de la cellule hôte précédente.
4) L'ARN est déplacée et dégradée par une activité RNase H. La
transcriptase inverse agit maintenant comme une ADN polymérase
ADN-dépendante et copie le nouvel ADN brin (-) en une copie ADN double
brin. Cette forme de l'ADN du virus est appelée un provirus.
5) L'ADN proviral double brin est inséré dans l'ADN de la cellule hôte (voir
ci-après) à l'aide d'une intégrase virale incorporée dans la particule
virale. Cet ADN est copié à chaque fois que l'ADN cellulaire est copié.
A ce stade, au niveau moléculaire, le provirus est exactement comme un
gène cellulaire normal.
6) La totalité de l'ARN génomique brin (+) est transcrit à partir de
l'ADN proviral intégré par l'ARN polymérase II de l'hôte. L'ARN
génomique est coiffé et polyadenylé, tout comme un ARNm cellulaire.
Cet ARN génomique complète sert également d’ARNm codant pour les
polyprotéines Gag et Pol.
L'ARN génomique est épissé par des enzymes nucléaires de l'hôte pour donner
les ARNm des autres protéines virales telles que Env. L'ARN de certains
rétrovirus plus complexes tels que le HTLV-1 et le VIH subit un épissage
multiple (voir chapitre 7, VIH).
Un avantage de ce mode de réplication est qu'il permet la réplication du
virus dans des cellules différenciées qui ne se divisent plus, puisque la
seule polymérase de la cellule hôte usurpée par le virus est l’ARN
polymérase II, qui est présente dans toutes les cellules.
|
|
|
|
|
L'utilisation de l'ARN polymérase II
de l'hôte pour recopier le génome viral à partir de l'ADN proviral entraine
des problèmes majeurs pour le virus, dont:
1) l'ARN polymérase II ne copie
pas les séquences de régulation en amont et en aval des gènes. Il ne
fait que copier les informations nécessaires pour fabriquer des
protéines virales.
2) L'absence de correction
d'erreurs de copie par l'ARN polymérase II.
L'ARN polymérase II ne transcrit
pas tout l'ADN proviral
Le problème vient du fait que, lors
de la transcription des gènes, l'ARN polymérase II a besoin de sites de
reconnaissance et de contrôle en amont du site d'initiation de
transcription. Le site en amont ou se fixe la molécule de la polymérase est
appelé le promoteur. Les promoteurs ne sont pas eux-mêmes copiés en ARNm,
car après sa liaison au promoteur, la polymérase commence la transcription à
un site en aval du promoteur. La polymérase continue à transcrire
l'ADN en ARN jusqu'à ce qu'il atteigne un signal de terminaison /
polyadenylation. Les séquences de l'ADN proviral en aval du site de
polyadenylation ne sont pas copiées par l'ARN polymérase II. De plus, en
amont du promoteur proviral se trouvent des séquences de contrôle qui
modulent la transcription du gène. Elles sont appelées des enhanceurs. Ce
sont des éléments essentiels de tout gène qui doivent être présents à
proximité du promoteur pour que l'ARN polymérase II puisse initier la
transcription. Ces séquences ne sont pas copiées en ARN
En effet, l'ARN polymérase II de la
cellule hôte a pour fonction de produire de l'ARN messager, qui est une
copie jetable d'une partie seulement du génome de la cellule. Pour fabriquer
une protéine, la molécule d'ARNm n'a pas besoin des séquences de contrôle du
gène d'origine. Ainsi, l'utilisation de l'ARN polymérase II de l'hôte
devrait avoir comme conséquence que les séquences de contrôle dans l'ADN
proviral d'origine ne sont pas recopiées dans l'ARN génomique du virus.
Cela signifie que soit l'ADN proviral
du virus doit s'intégrer dans l'ADN de l'hôte en aval d'un promoteur, et en
amont des sites de terminaison d'un gène de l'hôte – qui statistiquement
doit être quasi-impossible – ou alors, il doit trouver un moyen de
fournir ses propres séquences de contrôle, malgré le fait que, comme nous
venons de voir, ces séquences ne sont pas recopiés dans l'ARN génomique par
l'ARN polymeras II de l'hôte. En effet, le virus réussit cette deuxième
option par un tour de passe-passe moléculaire aussi élégant que complexe.
Comment un rétrovirus peut-il
fournir ses séquences régulatrices de la transcription si elles ne sont pas
transcrites lorsque l'ADN proviral est copié sous forme d'ARN génomique?
Voici un résumé bref et très
incomplet de la façon dont un rétrovirus réussit à copier ses séquences
régulatrices à chaque cycle de réplication virale. Ce processus est
également illustré par une
animation Flash.
|
| |
1) L'ARN viral est composé de trois régions. A
chaque extrémité sont des séquences répétées (appelées, sans surprise, des
répétitions terminales). Les séquences répétées (R) (en vert sur la figure
15) ne codent pas pour des protéines. Entre les deux répétitions, il y a une
région unique (non répétée) qui comprend les gènes viraux codant pour les
polyprotéines (gag, pol et env) ainsi que d'autres séquences uniques à
chaque extrémité qui ne codent pas pour des protéines. Près de l'extrémité 5
'du génome se trouve est la région U5 (pour séquence Unique 5') et près de
l'extrémité 3' est la région U3 (Unique 3'). La séquence PBS ("Primer
Binding Site" Figure 5) est le site de liaison des amorces. L'ARNt se lie
ici et permet à transcriptase inverse d'initier la synthèse de l'ADN à
partir l'ARN génomique du virus. La PPT (PolyPurine Tract) est une séquence
polypurinique (nucléotides A et G).
2) L'ADN proviral du virus est plus compliqué.
Nous constatons que la région U3 de l'ARN génomique a été copiée et
transposée à l'extrémité opposée du génome. Inversement, la région U5 a été
copiée et transposée à l'autre extrémité. Cela donne à l'ADN intégré la
structure représentée dans la Figure 5B. Pour plus de commodité, un seul
brin de l'ADN est affiché.
|
| |
La rétrotranscription de l'ARN viral en ADN proviral s'effectue
comme suit :
1) Début de la transcription inverse. La synthèse de l'ADN
démarre à l'ARNt situé au PBS, et continue jusqu'à l'extremité
5' de l'ARN viral. Ce premier brin d'ADN complémentaire comprend
donc les séquences complémentaire aux régions U5 et R de l'ARN
génomique. L'activité RNase H de la transcriptase inverse (qui
détruit l'ARN uniquement quand celui-ci fait partie d'un
hétéroduplexe avec de l'ADN) dégrade les régions U5 et R à
l'extrémité 5' de l'ARN viral.
2) Premier transfert de brin. L'ADN complémentaire à la séquence
R est désormais simple brin, et peut s'hybrider avec la séquence
R qui se trouve à l'extrémité 3' de l'ARN génomique. La
transcription inverse redémarre, et cette fois-ci, la totalité
de l'ARN génomique peut être recopiée, jusqu'à la séquence PBS
(car les séquences U5 et R ont été détruites précédemment par
l'activité RNase H). Ensuite, l'activité RNase H de la
transcriptase inverse dégrade l'ARN génomique qui vient d'être
recopié en ADN, à l'exception de la région PPT, qui résiste à
son activité enzymatique.
3) Début de la synthèse du deuxième brin. L'ARN de la séquence
PPT est utilisé comme amorce pour démarrer la synthèse du
deuxième brin de l'ADN proviral. Ce deuxième brin comprend les
séquences U3, R et U5. La séquence PBS est également copiée, car
l'amorce en ARNt forme toujours l'extrémité 5' du premier brin
d'ADNc, créant une région d'hétéroduplexe ARN-ADN qui sera par
la suite dégradé par l'activité RNase H.
4) Deuxième transfert de brin. La région PBS du deuxième brin
d'ADNc se trouve désormais sous forme d'ADN simple brin, et peut
s'hybrider avec la séquence complémentaire au PBS qui forme
l'extrémité 3' du premier brin de l'ADNc. Ce transfert de brins
place la séquence U3-R-U5-PBS en position d'amorce pour la
synthèse du reste du deuxième brin d'ADNc. En même temps, le
premier brin d'ADNc sera complété, en utilisant les séquences
U3-R-U5 comme matrice.
Par ce processus quelque peu complexe, l'ARN simple brin du
génome viral est copié en ADN double brin qui possède des
répétitions terminales plus grandes, car les régions U3 et U5 sont
aussi présentes à chaque extrémité du génome. Les régions U3-R-U5
sont connues comme les longues répétitions terminales ou LTR. La
région U3 contient toutes les séquences du promoteur qui sont
nécessaires pour démarrer la transcription de l'ARN viral au début
de la région R, tandis que la région U5 contient toutes les
informations nécessaires pour terminer la transcription après
l'autre séquence R. En outre, les LTR contiennent de l'information
qui améliore le degré de transcription des trois gènes rétroviraux (régions
dites "Enhanceur"). Ces activateurs se trouvent principalement en
amont de la région promoteur en U3, mais peuvent comprendre des
éléments qui se chevauchent avec la partie codante du génome viral.
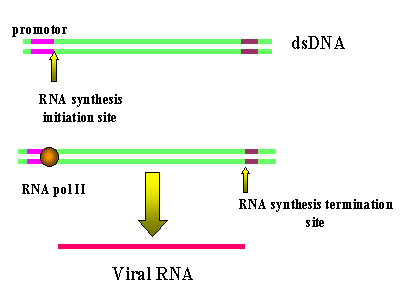
L'ARN polymérase II de l'hôte copie de l'ADN proviral en ARN
génomique. Puisque le site d'initiation de la polymérase se trouve
après le promoteur en U3, la transcription commence exactement au
début de la région R (Figure 6). Ainsi, nous obtenons un copie
fidèle (ou presque - voir ci-dessous) de l'ARN qui est entré dans la
cellule. Les séquences de terminaison et de polyadenylation sont
comprises dans la région U5, qui également n’est pas copié en ARN.
|
|
|
|
A
 5A Structure du génome rétroviral en ARN simple brin qui
se trouve à l'intérieur de la particule virale
5A Structure du génome rétroviral en ARN simple brin qui
se trouve à l'intérieur de la particule virale
B
 5B Structure d'un génome proviral d'un rétrovirus, en ADN double-brin
intégré dans le génome de la cellule hôte.
5B Structure d'un génome proviral d'un rétrovirus, en ADN double-brin
intégré dans le génome de la cellule hôte.
Figure 5
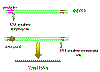 Figure 6
Les LTRs sont perdues lors de la transcription de l'ADN proviral à
partir du LTR par l'ARN polymérase II.
Animated version
here
Figure 6
Les LTRs sont perdues lors de la transcription de l'ADN proviral à
partir du LTR par l'ARN polymérase II.
Animated version
here |
En raison de ce mécanisme, il peut y avoir qu'un seul site de
promoteur (à partir de la région U3) pour les trois gènes viraux. Ils
doivent donc être transcrits tous ensemble, et la machinerie d'épissage de
la cellule hôte prend en charge le transcrit primaire pour former les
différents ARNm (notamment l'ARNm codant pour les glycoprotéines de
l'enveloppe). (Voir le chapitre 7, traitant du VIH, chez lequel les
mécanismes précis de contrôle d'expression génique ont été bien élucidés).
Contrairement à la situation chez des virus oncogénes à ADN, il n'y a pas de
distinction entre les gènes précoces et tardifs.
On peut se demander pourquoi, si la région U5 contient les séquences de
terminaison de transcription et des sites de polyadenylation, la
transcription ne se termine pas tout simplement à la fin de la première
région R de la LTR (Figure 5b) sans jamais entrer dans les régions du génome
codant pour les protéines virales. La fonction du site de terminaison de la
première séquence U5 est supprimée, souvent par un mécanisme complexe
impliquant la structure secondaire de l'ARN qui vient d'être transcrit. Chez
certains rétrovirus une séquence spécifique dans le gène gag fournit le
contexte pour supprimer l'activité de la fin de la première U5. Clairement
la seconde séquence U5 n'a pas de gène gag suivant, et la transcription est
donc terminée.
Cette stratégie de réplication dans lequel l'ARN viral est d'abord copié en
ADN (par la transcriptase inverse) qui ensuite sert de matrice pour la
production des ARNm et des protéines virales pose un autre problème pour le
virus. La première étape (ARN à l'ADN) est réalisée par une enzyme virale
qui n'est pas normalement dans la cellule. Pourtant, cette étape de
transcription doit avoir lieu avant que la transcription de l'ARNm ou la
traduction des protéines virales peuvent se produire. Afin de résoudre ce
problème, le virus transporte une dizaine de copies de la transcriptase
inverse dans la cellule avec elle. Celles-ci ont été incorporées lorsque le
virus a été assemblé dans la cellule hôte précédente. En théorie, l'ARN
génomique du virus rentrant dans la cellule pourrait agir comme un ARNm,
mais en réalité son association avec la nucléocapside et d'autres protéines
virales l'en empêche. Ainsi, aucune expression des gènes viraux n'est
possible avant l'intégration de l'ADN proviral dans le génome de la cellule
hôte, ce qui nécessite l'activité de la transcriptase inverse et de
l'intégrase virales.
|
|
 Structure génomique d'un rétrovirus typique et d'un rétrovirus porteur
d'un oncogène viral (Virus de Sarcome de Rous)
Structure génomique d'un rétrovirus typique et d'un rétrovirus porteur
d'un oncogène viral (Virus de Sarcome de Rous)Figure
7
 Certains rétrovirus transformant portent un oncogène à la place de l'un
de leur gènes essentiels
Certains rétrovirus transformant portent un oncogène à la place de l'un
de leur gènes essentiels
Figure 8 |
La structure présentée dans la Figure
5a et la partie supérieure de la Figure 7 montre le génome d'un rétrovirus
typique comportant les gènes de gag, pol et env dont
aucun des trois n'est oncogène. Si le virus devra transformer une cellule
normale en cellule cancéreuse, il doit comporter un gène, en plus des gènes
gag, pol et env qui est capable de perturber le
contrôle du cycle cellulaire et d'induire le phénotype transformé. On doit
donc trouver un oncogène viral chez les rétrovirus qui sont capable
de transformer le phénotype de chaque cellule infecté vers celui d'une
cellule néoplasique. Il est important d'insister sur le fait que cet
oncogène n'est pas nécessaire pour la réplication du virus – il s'agit d'un
gène supplémentaire qui confère au virus la capacité de transformer la
cellule hôte.
Le premier oncogène de ce type a été
identifié chez le virus de sarcome de Rous (RSV). Puisque ce virus induit
des sarcomes, l'oncogène fut appelé src. Le RSV possède un gènome
gag/pol/env complète, suivi de l'oncogène src
(Figure 7). La délétion ou des mutations dans le gène src ne
modifient pas la réplication du virus, mais rendent le virus incapable
d'induire des tumeurs. Le RSV est un exemple unique parmi les rétrovirus à
fort potentiel transformant, en ce sens que l'incorporation de l'oncogène
src dans son génome n'a pas perturbé les gènes gag, pol et
env qui sont essentielles pour la réplication virale.
Chez les autres rétrovirus à fort
potentiel transformant, une partie du génome viral a été remplacée par
l'oncogène (Figure 8). Il s'ensuit deux conséquences:
1)
La protéine encodée par l'oncogène est souvent exprimée comme une
protéine de fusion avec une partie d'un gène viral.
2)
Les rétrovirus ayant incorporé un oncogène sont défectueux, et ne
sont pas capables de se répliquer de façon autonome. Ils ont donc besoin
d'une co-infection par un virus auxiliaire (le rétrovirus "wild-type" qui ne
possède pas l'oncogène).
Une quarantaine d'oncogènes ont été
identifié par l'étude de rétrovirus transformants. Chacun est désigné par un
code à trois lettre, qui le plus souvent est dérivé du nom du virus chez
lequel l'oncogène a été décrit. Puisqu'il s'agit d'oncogènes présents dans
des génomes viraux, le nom de l'oncogène
est précédé
par un "v". Quelques exemples sont:
|
Virus |
Oncogène |
| Rous sarcoma virus |
v-src |
| Simian sarcoma
virus |
v-sis |
| Avian
erythroblastosis virus |
v-erbA ou v-erbB |
| Kirsten murine
sarcoma virus |
v-Kras |
| Moloney murine
sarcoma virus |
v-mos |
| MC29 avian
myelocytoma virus |
v-myc |
|
| |
|
| |
Les proto-oncogènes sont
présents chez les cellules non-transforméesSuite à
l'identification d'oncogènes viraux, une découverte étonnante (récompensée
par le Prix Nobel de Médicine en 1989) a été faite – la présence
d'homologues des oncogènes rétroviraux dans l'ADN des cellules non-cancéreuses,
qui n'avaient pas été infectées par un rétrovirus. Ces homologues
sont souvent impliqués dans le contrôle de la croissance et de la
différenciation cellulaires. Certains peuvent provoquer le
développement des cancers sous certaines conditions (par exemple,
quand ils sont surexprimés). Les homologues cellulaires des
oncogènes rétroviraux sont appelés des "proto-oncogènes". Afin de
distinguer les oncogènes viraux de leurs homologues cellulaires, les
deux types de gène sont désignés comme de "v-onc" et des "c-onc",
respectivement. Les v-onc sont d'origine cellulaire (et non pas
l'inverse – c'est à dire que les c-onc ne sont pas d'origine viral).
Il est probable que les rétrovirus transformants ont incorporé toute
ou partie d'un c-onc dans leur génome, qui par la suite a été
modifié lors des multiples cycles de réplication du virus.
Caractéristiques des proto-oncogènes
cellulaires
1) Ce sont des gènes cellulaires typiques, qui comme la
plupart des gènes eucaryotes sont composés de plusieurs exons et
introns. (tandis que les oncogènes rétroviraux – les v-onc – ne
possèdent pas d'introns)
2) Ils sont transmis de génération en génération de façon
Mendélienne, car il s'agit de gènes cellulaires "normaux", qui
remplissent des fonctions cellulaires essentielles.
3) Comme pour tous les gènes d'un génome eucaryote, ils se
trouvent toujours au même locus dans le génome. (pour
comparaison; à quoi pouvait-on s'attendre si des rétrovirus
porteurs des v-onc s'étaient intégrés dans le génome cellulaire
?)
4) Ils ne sont pas associés aux séquences LTR (les v-onc sont
toujours entourés des LTR)
5) Les oncogènes rétroviraux ressemblent les c-onc de leur
espèce hôte. Par exemple, le v-src du RSV est plus proche du c-src
du poulet que du c-src humain.
6) Les oncogènes cellulaires sont exprimés à un moment précis
dans la vie de la cellule – lorsqu'elle se divise et se
différencie, par exemple, mais ne sont pas exprimés de façon
constante dans une cellule non-cancéreuse. Les protéines
exprimées à partir des c-onc sont généralement impliquées dans
le contrôle de la croissance et du cycle cellulaire.
7) Les oncogènes cellulaires sont fortement conservés.
Dès lors que l'on se rend compte des ressemblances entre les c-onc
et les v-onc, une question qui se pose est la suivante: pourquoi les
v-onc provoquent-t’ils un tel dérèglement de la croissance
cellulaire? La réponse se trouve dans les différences entre les v-onc
et les c-onc, en particulier les mutations qui se sont accumulées
chez les v-onc pendant, ou à la suite de leur incorporation dans le
génome rétroviral. Par exemple:
Des substitutions ou délétions dans la séquence des v-onc qui
résultent en la traduction d'une protéine avec une fonction modifiée.
Les v-onc sont souvent exprimés sous forme de protéines de fusion
avec un gène viral. Encore une fois, la fonction d'un tel v-onc sera
modifiée par rapport au c-onc.
L'expression des v-onc est sous le contrôle du LTR rétroviral, et
non pas sous le contrôle des séquences régulatrices de son c-onc
homologue. Ceci résulte en la surexpression du v-onc.
|
 Oncogénèse par insertion du promoteur. La LTR agit comme promoteur,
induisant une expression abérrante d'un oncogène cellulaire.
Oncogénèse par insertion du promoteur. La LTR agit comme promoteur,
induisant une expression abérrante d'un oncogène cellulaire.
Figure 9
 Oncogénèse par insertion d'enhanceur. L'oncogène cellulaire est exprimé
à partir de son propre promoteur, mais le niveau d'expression est
augmenté par l'action enhanceur de la LTR rétrovirale.
Oncogénèse par insertion d'enhanceur. L'oncogène cellulaire est exprimé
à partir de son propre promoteur, mais le niveau d'expression est
augmenté par l'action enhanceur de la LTR rétrovirale.
Figure 10
A
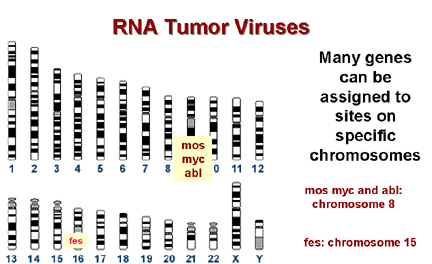
 Les oncogènes cellulaires se trouvent à des
sites spécifiques sur l'ADN génomique.
Les oncogènes cellulaires se trouvent à des
sites spécifiques sur l'ADN génomique.
B
 De nombreux sites de cassure chromosomique observés dans les
cellules cancéreuses se trouvent à proximité d'un oncogène cellulaire.
De nombreux sites de cassure chromosomique observés dans les
cellules cancéreuses se trouvent à proximité d'un oncogène cellulaire.
Figure 11
|
Les rétrovirus à faible
potentiel transformant ne portent pas de v-onc
Parmi les rétrovirus transformant, deux types de virus se
distinguent. Les rétrovirus à fort potentiel transformant - comme le
RSV - qui induisent des tumeurs rapidement chez presque 100% des
animaux infectés (d'où le nom, "acutely-transforming retroviruses"),
et les rétrovirus à faible potentiel transformant - comme par
exemple, le Avian Leukosis Virus (ALV) - qui induisent des tumeurs
chez une proportion plus faible des animaux infectés, après un laps
de temps plus long (d'où le nom, "chronically-transforming
retroviruses"). Ces derniers ne possèdent pas d'oncogène viral, et
sont dotés uniquement des gènes rétroviraux gag/pol/env standards.
Comment font les rétrovirus à faible potentiel transformant pour
induire des tumeurs en l'absence d'un oncogène viral?
Comme tous les rétrovirus, l'ALV s’intègre dans le génome de la
cellule hôte de façon aléatoire. En revanche, chez les cellules
tumorales, le génome proviral se trouve toujours à proximité d'un
oncogène cellulaire appelé c-myc. Ce qui semble se passer, est que
la très grande majorité des cellules infectées par l'ALV ne sont pas
transformées par le virus, mais dans des rares cas, lorsque le virus
s'intègre à proximité du gène c-myc, la cellule infectée est
transformée. La croissance de ce rare clone cellulaire forme la
tumeur. Le niveau d'expression du c-myc est beaucoup plus élevée
chez les cellules transformées que chez les cellules non-infectées.
L'insertion du provirus d'ALV à proximité du gène c-myc peut
induire la surexpression de ce dernier par deux mécanismes.
1) Lorsque le provirus s'intègre en amont du gène c-myc, le
promoteur du LTR induit la transcription constitutive du gène c-myc.
Ce mécanisme est appelé transformation par insertion du
promoteur (Figure 9).
2) Dans certaines tumeurs, l'ADN proviral se trouve en aval
du gène c-myc. Dans ce cas, les éléments enhanceurs dans la LTR
proviral augmente le niveau de transcription à partir du
promoteur autologue du gène c-myc. Ce mécanisme est appelé
transformation par insertion d'enhanceur (Figure 10).
Pourquoi l'insertion à proximité du gène c-myc est-elle si
importante? La protéine encodée par le gène c-myc est localisée dans
le noyau, et joue un rôle dans le contrôle de la synthèse de l'ADN.
La surexpression du c-myc (indépendamment d'une infection par l’ALV)
mène à réplication incontrôlée de l'ADN.
L'implication des oncogènes
cellulaires dans des cancers non-induits par des virus
L'identification des oncogènes viraux chez les rétrovirus a mené
à la découverte des gènes homologues (appelé proto-oncogènes) qui se
trouvent dans toutes les cellules. Normalement, l'expression des
proto-oncogènes cellulaires est strictement contrôlée par la
cellule, car ils sont impliqués dans la division cellulaire.
Toutefois, nous venons de voir que l'infection par un rétrovirus
peut induire le cancer de deux façons: soit il peut porter un
oncogène viral dans la cellule, soit l'intégration de l'ADN proviral
peut activer l'expression d'un proto-oncogène cellulaire.
Est-il possible qu'un dérèglement de l'expression ou de la
fonction des c-onc puisse provoquer le cancer en l'absence d'une
infection virale? La réponse est oui. Des remaniements
chromosomiques peuvent transférer un c-onc sous le contrôle d'un
autre promoteur/enhanceur, menant à la surexpression de l'oncogène,
ou menant à l'expression d'une protéine de fusion entre le c-onc et
un autre gène. Des mutations ponctuelles (induits par exemple, par
des mutagènes chimiques, comme le tabac) dans des c-onc peuvent
également modifier leur fonction.
Oncogènes cellulaires et
remaniements chromosomiques
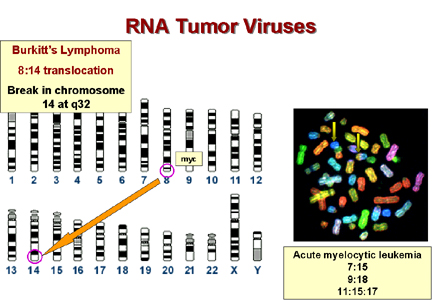
En ce qui concerne les remaniements chromosomiques, de nombreuses
tumeurs sont caractérisées par un caryotype aberrant, comprenant en
particulier des translocations chromosomiques (transfert d'un
segment chromosomique à un bras d'un autre chromosome). Dans de
nombreux cas, les sites de translocations dans les cellules
tumorales sont à proximité d'un c-onc (Figure 11). Etant donné le
nombre relativement restreint de c-onc dans le génome humain, cette
distribution de translocations chez les cellules tumorales ne peut
pas être dû au hasard.
|
Cancer |
c-onc |
Remaniement chromosomique |
| Lymphome de Burkitt* |
Myc |
t(8:14) |
| Leucémie aigüe
myéloblastique |
AML1-ETO |
t(8:21) |
| Leucémie aigüe
lymphoblastique B |
BCR-abl |
t(9:22) |
| Leucémie myéloïde
chronique |
BCR-abl |
t(9:22) |
| Leucémie aigüe
promyélocytaire |
PML-RAR |
t(15:17) |
| Cancer de l'ovaire |
myb |
t(6:14) |
* Dans le cas du lymphome de Burkitt le gène c-myc sur le
chromosome 8 est ramené à un site au chromosome 14 à proximité du
gène de la chaîne lourde des immunoglobulines. Le gène c-myc se
trouve donc sous le contrôle du promoteur de ce dernier, qui est
très actif chez les lymphocytes B. Cette tumeur est donc une
transformation maligne des lymphocytes B.
De plus, le lymphome de Burkitt est associé à l'infection par le
virus Epstein-Barr, un -Herpesvirus qui infecte les lymphocytes B.
Les Herpesvirus provoquent des remaniements chromosomiques chez les
cellules infectées, et si l'un des remaniements résulte en la
translocation 8:14, le gène c-myc se trouvera à proximité du
promoteur du gène de la chaîne lourde des immunoglobulines, qui est
justement actif chez les cellules infectées par le virus
Epstein-Barr.
|
.
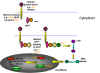 Différentes façons par lesquelles la
modification des proto-oncogènes cellulaires peuvent induire la
transformation cellulaire.
Différentes façons par lesquelles la
modification des proto-oncogènes cellulaires peuvent induire la
transformation cellulaire.
Figure 12
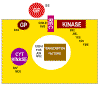
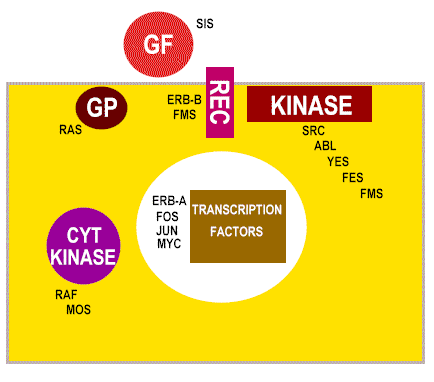
Les différentes classes de protéine codées par
des proto-oncogène cellulaires
GF = Facteurs de croissance (Growth Factors)
REC = Récepteurs membranaires
GP = Transducteurs de signal à protéine-G
KINASE = Tyrosine kinases membranaires
CYT KINASE = Kinases cytoplasmiques
Figure 13
|
Un exemple de mutations
ponctuelles qui activent un c-onc lors du développement du cancer
concerne le proto-oncogène K-Ras, qui a initialement été
identifié comme un v-onc dans le génome de la souche Kirsten du
virus de sarcome murin. Dans environ 90% des carcinomes du pancréas, le
gène K-Ras est muté chez les cellules tumorales. La plupart du
temps la mutation implique la substitution d'un valine à la position 12
de la protéine à la place du glycine, qui est présent dans la protéine
sauvage. Cette mutation ponctuelle rend la protéine constitutivement
active, et induit la prolifération cellulaire. Les mutations ponctuelles
dans les différents proto-oncogènes de la famille Ras (K-Ras, H-
Ras, et N- Ras) sont présentes dans 20 à 30% des cancers
humains, tous types de tumeur confondus.
Comme indiqué ci-dessus, les c-onc
sont des gènes cellulaires normaux qui remplissent des fonctions
essentielles dans le contrôle du cycle cellulaire. Ils sont impliqués
dans la synthèse de l'ADN et les voies de signalisation qui mènent à la
mitose et à la prolifération cellulaire. Les c-onc peuvent être
divisés en deux groupes: ceux qui codent pour des protéines nucléaires,
et ceux qui codent pour des protéines extra-nucléaires. Ces derniers
sont, pour la plupart, associés à la membrane plasmique (Figures 12 et
13)
Oncogènes cellulaires
codant pour des protéines nucléaires:
eg. myc, myb. Ces protéines sont soit des facteurs
de transcription, soit des protéines directement impliquées dans
le contrôle de la réplication de l'ADN. Le néoplasie est associé
avec une expression constitutive de ce type d'oncogène, tandis
que l'expression de ces gènes chez la cellule normale est
strictement contrôlée. En revanche, un fort niveau d'expression
n'est pas toujours nécessaire.
Oncogènes cellulaires
codant pour des protéines cytoplasmiques ou associées à la
membrane plasmique: eg. abl,
src, ras, erbB. Dans des tumeurs ce type
d'oncogène ne montre pas de modification du niveau d'expression
– par exemple, la surexpression de la forme sauvage du src
n'induit pas la transformation cellulaire. En revanche, les
formes mutées de ces protéines (que ce soient des v-onc,
ou des c-onc ayant été touchés par des mutations
ponctuelles) sont constitutivement actives, tandis que les
formes normales de ces c-onc sont actives de façon
ponctuelle. Si l'on considère les proto-oncogènes de ce groupe
comme des interrupteurs moléculaires, les formes mutées sont des
interrupteurs qui restent coincées sur la position "On".
Dans les deux cas, la
mutation dans un c-onc est dominante.
Par exemple, si un allèle du gène erb-B (un homologue du
récepteur du facteur de croissance EGF) incorpore une mutation qui
le rend constitutivement actif (c'est à dire que son activité de
tyrosine kinase n'est pas dépendant de la présence de l'EGF), la
prolifération cellulaire sera engagée en permanence, même si le
deuxième allèle est normal.
|
Fonction de la
protéine codée par le proto-oncogène cellulaire |
Exemples |
|
Facteur de transcription (nucléaire) |
Myc |
|
Facteur de croissance (secrétée) |
sis
: le v-onc sis est une forme modifiée de
la chaîne B du PDGF (Platelet Derived Growth Factor) |
|
Récepteurs des facteurs de croissance |
erb-B
: récepteur du facteur de croissance EGF (Epidermal
Growth Factor)
fms
: récepteur du facteur de croissance M-CSF (Macrophage
Colony Stimulating Factor)
|
|
Signalisation à la face interne de la membrane de la
fixation d'un facteur de croissance |
src
: tyrosine kinase membranaire |
|
Protéines à activité GTPase impliquées dans la
transduction du signal de la membrane cellulaire au
noyau |
Ras |
|
 Le flux d'information chez un virus à ADN est semblable à
celui chez une cellule eucaryote.
Le flux d'information chez un virus à ADN est semblable à
celui chez une cellule eucaryote.
Figure 14 |
La découverte des oncogènes cellulaires a ouvert la voie à
l'élucidation des mécanismes par lesquels les cancers qui ne sont pas
induits par des virus peuvent se produire.
Nous examinerons les fonctions des produits protéiques des oncogènes
viraux et cellulaires dans la cellule infectée et dans les cellules où
les proto-oncogènes cellulaires sont exprimés. Nous verrons que leurs
fonctions suggèrent fortement des mécanismes par lesquels les cellules
peuvent être transformées en un phénotype néoplasique. La découverte des
oncogènes cellulaires a conduit à la découverte d'une autre classe de
gènes cellulaires, les gènes suppresseurs de tumeur ou anti-oncogènes.
Initialement, l'implication des oncogènes viraux et cellulaires dans les
tumeurs provoquées par des rétrovirus était beaucoup plus apparente que
l'implication des oncogènes portés par des virus à ADN, mais la
découverte de gènes suppresseurs de tumeurs a conduit à l'élucidation du
mode d'action des oncogènes chez les virus à ADN. Il convient de noter
que, bien que les rétrovirus aient contribué à l'élucidation des
mécanismes de la carcinogénèse, l'immense majorité des cancers humains
ne sont pas le résultat d'une infection rétrovirale (bien que les
rétrovirus représentent une cause importante de cancers chez certains
animaux). Les virus à ADN, en revanche, sont associés à plusieurs types
de cancer humain.
Les virus transformants à ADN et la
découverte des anti-oncogènes cellulaires
Les premiers
virus à ADN associés à des cancers étaient le virus du fibrome du lapin, et
le virus du papillome de Shope, tous les deux découverts par Richard Shope
dans les années 1930. Les papillomes sont des tumeurs bénignes, comme les
verrues, des cellules épithéliales. Richard Shope a découvert ces deux virus
en préparant un extrait filtré d'une tumeur à partir d'un lapin de garenne,
qu'il a injecté à un autre lapin, chez lequel un papillome bénin a développé.
Toutefois, lorsque le filtrat a été injecté dans un lapin domestique, le
résultat était un carcinome, qui est une tumeur maligne. Une observation
importante était qu'il n'était plus possible d'isoler le virus infectieux à
partir de la tumeur maligne parce que le génome du virus s'était intégré
dans les chromosomes des cellules malignes.
Les virus transformant à ADN possèdent un génome en ADN qui est
transcrit en ARNm par la polymérase de la cellule hôte, qui est ensuite
traduit en protéines (Figure 14). Ces virus ont deux modes d'infection :
1: Dans les cellules permissives, toutes les parties du génome
viral sont exprimés. Cela conduit à la réplication virale, la lyse
cellulaire et la mort cellulaire
2: Dans les cellules qui sont non permissives pour la réplication
virale, de l'ADN viral peut être intégré à des sites aléatoires dans
les chromosomes de la cellule hôte. Seule la partie du génome viral
comportant les gènes précoces (par exemple, l'antigène T des
Polyomaviridae) est exprimée. Les protéines structurales du virus ne
sont pas produites et aucune particule virale n’est libérée.
|
 Polyomavirus SV40 en microscopie électronique à
transmission. Dr Erskine Palmer CDC.
Polyomavirus SV40 en microscopie électronique à
transmission. Dr Erskine Palmer CDC.
Figure 15A
 Figure 15B
Figure 15B
Human polyomaviruses and associated diseases.
The organs to which each human polyomavirus has tropism and causes
disease.
doi:10.1371/journal.ppat.1003206.g001
From: The Rapidly Expanding Family of Human Polyomaviruses: Recent
Developments in Understanding Their Life Cycle and Role in Human
Pathology. Martyn K. White, Jennifer Gordon and Kamel Khalili.
PLOS Pathogens. Used under Creative Commons License |
Les virus transformants à ADN dans
des systèmes modèles
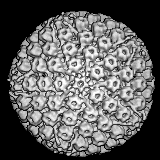
Polyomaviridae Les
Polyomaviridae sont de petits virus à ADN icosaédriques non enveloppés
(Figure 15). La protéine majeure de capside, VP1, est présente sous
forme de 72 pentamères dont chaque pentamère est associé à une molécule
d'une protéine de capside mineure, soit VP2 ou VP3. L'ADN à l'intérieur
du virion est complexé avec des protéines histones codées par la cellule
hôte.
Les Polyomaviridae ont un génome en ADN double brin circulaire d'environ
5 kilobases de longueur pour au moins 2 protéines précoces (les
antigènes "T" et "t") et les protéines tardives formant la capside (VP1,
VP2/3).
Le Virus Polyoma murin
Le virus polyoma murin a été ainsi nommée parce qu'il provoque la
formation de tumeurs diverses chez différentes espèces animales, à
de nombreux sites différents. Il a été initialement isolé de la
souche de souris AK, et il se réplique de façon efficace dans les
cellules de souris. Il provoque des leucémies chez les souris et les
hamsters. Le virus SV40
Le virus SV40 (Figure 15) a été découvert dans les cellules de rein
de singe rhésus utilisées pour la culture du virus de la
poliomyélite lors de la fabrication du vaccin inactivé Salk. Il a
été constaté que lorsque le virus de la poliomyélite inactivé a été
ajouté à cellules de rein de singe vert, le vaccin provoquait un
effet cytopathogène (CPE) indicatif de la présence d'un virus
infectieux qui n'avait pas été inactivé par le formol utilisé lors
de la production du vaccin. Le virus SV40 se réplique dans les
cellules de rein de singe rhésus, mais ne provoque pas de CPE sur ce
type cellulaire, ce qui explique pourquoi ce contaminant n'avait pas
été détecté dès le début de production du vaccin.
La présence d'anticorps spécifiques de l'antigène T du virus SV40
chez un grand nombre des premiers enfants ayant reçu le vaccin Salk
contre la poliomyélite a indiqué que ces enfants avaient été
contaminés par le virus SV40. Aucune incidence élevée du cancer n’a
été trouvé chez ces personnes, démontrant que ce virus n'est pas
associé avec le développement de cancer chez l'Homme.
Bien que le virus SV40 soit un virus de singe qui n'a pas d'effet
apparent sur son hôte naturel, il provoque les sarcomes lorsqu'il
est injecté à des hamsters jeunes. Les cellules tumorales de hamster
ne produisent pas de virus infectieux. Les Polyomaviridae
humains
Les deux premières souches de Polyomavirus humains, connus sous le
nom BK et JC ont été isolées en 1971. La souche BK a été isolée à
partir de l'urine d'un patient greffé du rein, et la souche JC a été
isolée à partir du cerveau d'un patient atteint de lymphome de
Hodgkin, qui a progressé vers une leuco encéphalopathie multifocale
progressive (LEMP). Les virus BK et JC provoquent des tumeurs
lorsqu'ils sont injectés dans les animaux, bien qu’aucun des deux
virus ne provienne d'une tumeur. 70 à 80% de la population humaine
est séropositif pour virus JC. Ce virus est connu pour être la cause
de la LEMP, une maladie associée à l'immunosuppression. En 1979,
l'incidence de cette maladie était de 1,5 pour 10 millions
d'habitants. Il est devenu beaucoup plus fréquent à cause de
l'épidémie du SIDA et est vu chez 5% des patients atteints d'un SIDA
clinique. Bien entendu, un traitement antirétroviral efficace permet
de maintenir les fonctions immunologiques chez les personnes
séropositives pour le VIH, et élimine donc le risque de survenu de
la LEMP.
Le virus BK est une cause importante de néphropathie menant à
l'échec de la greffe chez les transplantés rénaux sous traitement
immunosuppresseur. La quasi-totalité de la population dans les pays
occidentaux possède des anticorps anti-virus BK avant l'âge de 10
ans.
Trois autres virus polyoma humains ont été récemment décrit: les
virus KI, WU et le Polyomavirus à cellules de Merkel. Ce dernier
virus provoque un cancer rare de la peau (le carcinome à cellules de
Merkel, voir encadré).
Les Polyomaviridae sont généralement lytiques et lorsque la
transformation se produit, c'est parce que la cellule hôte est
non-permissive pour l'infection virale. Après l'intégration dans
l'ADN de l'hôte, seuls les gènes précoces sont transcrits en ARNm et
exprimés sous forme de protéine. Les protéines virales précoces sont
les antigènes tumoraux des Polyomaviridae. Parce que l'expression
des gènes codant pour les antigènes tumoraux est essentiel pour la
transformation des cellules, ils peuvent être classés comme des
oncogènes. Chez l'hôte naturel de ces virus, les cellules sont
permissives pour l'infection virale. Dans ce cas, même si les
oncogènes viraux sont exprimés chez la cellule infectée,
l'aboutissement du cycle de réplication viral implique la lyse de la
cellule, qui ne persiste donc pas dans un état transformé.
|
 Adenovirus © Dr Stephen Fuller 1998
Adenovirus © Dr Stephen Fuller 1998
 Adenovirus
en microscopie électronique à transmission. © Dr Linda M. Stannard 1995
(reproduit avec permission) Adenovirus
en microscopie électronique à transmission. © Dr Linda M. Stannard 1995
(reproduit avec permission)
Figure 16 |
Adenoviridae
Les Adenoviridae sont des virus non-enveloppés (Figure 16), ayant
des capsides d'un diamètre de 90nm – soit un peu plus grand que les
Polyomaviridae et les Papillomaviridae - et leur génome en ADN
double brin linéaire est d'environ 35 kilobases de longueur. Ils ont
été initialement isolés à partir d'amygdales et des végétations
adénoïdes humaines, et ils sont très oncogènes chez les animaux.
Chez les cellules tumorales, seule une partie du génome viral est
intégré dans l'ADN chromosomique de l'hôte. Cette partie comprend
plusieurs gènes précoces qui sont nécessaires pour la réplication de
l'ADN viral au cours de l'infection. Comme pour les Polyomaviridae,
l'induction de cancers par les Adenoviridae semble être la
conséquence d'une infection abortive chez les cellules qui ne sont
pas permissives pour la réplication virale. Chez l'hôte naturel des
adénovirus (c'est à dire Homo sapiens) la réplication mène à la lyse,
et non pas à la transformation maligne de la cellule infectée. Ainsi,
aucun cancer humain n'est associé à une infection par un adénovirus.
Les antigènes viraux
exprimés par les tumeurs sont des oncogènes
Les tumeurs provoqués par les Adenovirus ou des Polyomavirus
contiennent de l'ADN viral, mais ne produisent pas de virus
infectieux. Par contre, la présence des antigènes viraux induit la
formation des anticorps contre les protéines virales exprimées par
la tumeur. Dans le cas des Adénovirus, seule une partie du génome
viral est intégrée dans l'ADN génomique des cellules tumorales. Dans
le cas du virus SV40, même si le génome viral en entier peut
s'intégrer, seule la région codant pour l'expression des gènes
précoces du virus est exprimée par les cellules tumorales.
Les gènes précoces des petits virus à ADN (Polyomaviridae,
Papillomaviridae, et Adenoviridae) codent pour des protéines qui
préparent la cellule hôte pour la production des virus, et notamment
pour la réplication de l'ADN viral. Les gènes tardifs, en revanche,
sont impliqués dans la formation et la libération des particules
virales, et sont exprimés après la réplication de l'ADN viral.
Puisque les gènes précoces sont nécessaires pour la réplication de
l'ADN viral, il n'est peut-être pas surprenant qu'ils sont également
capables de stimuler la réplication de l'ADN de la cellule hôte.
Les protéines précoces du virus SV40 sont les protéines "T" et "t" (antigènes
"Grand T" et "Petit t"). L'antigène T stimule la réplication de
l'ADN viral en se fixant sur l'origine de réplication de l'ADN
viral, et en recrutant l'ADN polymérase au site d'initiation de
réplication virale. De plus, l'antigène T se fixe sur (et inactive)
les protéines cellulaires p53 et p105-Rb (pour Rétinoblastome) qui
jouent un rôle clé dans le contrôle de la réplication de l'ADN
cellulaire. Cette fonction de l'antigène T induit la transition de
la cellule hôte de la phase G0 vers la phase S du cycle cellulaire.
Les Polyomaviridae (comme la plupart des petits virus à ADN) doivent
induire l'entrée de la cellule dans la phase S car ils sont
dépendants de multiples facteurs cellulaires impliqués pour la
réplication de l'ADN viral.
En résumé, l'antigène T du virus SV40:
− est nécessaire pour la transformation maligne d'une cellule
− se fixent sur et inactive les protéines p53 et p105-Rb
− stimule la réplication de l'ADN virale et cellulaire
− se trouve majoritairement dans le noyau
− peut se fixer sur l'ADN cellulaire, si l'origine de réplication
virale est intégrée dans l'ADN cellulaire
L'antigène "petit t", quant à lui, inhibe l'activité enzymatique de
la famille de phosphatases PP2A. Etant donné que les phosphatases
PP2A sont des facteurs de régulation négatifs du cycle cellulaire,
l'inhibition de PP2A lève l'arrêt du cycle cellulaire.
Chez les tumeurs induites par les Adénovirus, la région du génome
viral intégrée dans l'ADN cellulaire code pour les protéines
précoces E1A et E1B. La protéine E1A interagit avec la protéine
cellulaire p105-Rb, tandis que la protéine E1B se fixe sur protéine
cellulaire P53, et l’inactive.
Ainsi, les mécanismes par lesquels les Polyomaviridae et les
Adenoviridae provoquent la transformation cellulaire se ressemblent.
Dans les deux cas, l'événement essentiel est l'intégration des gènes
précoces dans l'ADN de la cellule hôte, et l'expression constante de
ces gènes en l'absence de production des protéines tardives.
L’interaction entre les protéines précoces virales et les protéines
cellulaires jouant des rôles clés dans le contrôle de la réplication
de l'ADN et la division cellulaire induit la prolifération
cellulaire effrénée caractéristique d'une cellule cancéreuse. Ces
gènes précoces des virus à ADN sont donc des oncogènes viraux.
Il est important de souligner deux caractéristiques des oncogènes
des virus à ADN qui les distinguent des oncogènes rétroviraux:
− Ce sont des gènes véritablement viraux. Ils n'ont pas d'homologue
dans le génome de la cellule hôte.
− Ils remplissent des fonctions essentielles dans le cycle de
réplication viral. Ils sont notamment nécessaires pour permettre la
réplication de l'ADN viral, et un virus transformant à ADN chez qui
l'oncogène viral est délété ne peut pas se répliquer.
|
|

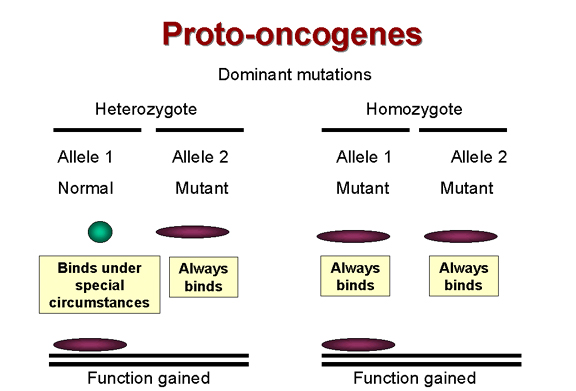
Des mutations dominantes
correspondent à une gain de fonction.
 Des mutations récessives correspondent à une perte de fonction.
Des mutations récessives correspondent à une perte de fonction.
Figure 17
 P105 RB et la protéine E1A de l'adénovirus
P105 RB et la protéine E1A de l'adénovirus
Figure 18
 P53 et son inactivation par le virus de l'hépatite C et
les papillomavirus
P53 et son inactivation par le virus de l'hépatite C et
les papillomavirus
Figure 19
|
Les anti-oncogènes (gènes
suppresseurs de tumeur)
Les virus transformants à ADN portent des
oncogènes comme l'antigène T du virus SV40, mais comment se fait-il que
ces gènes viraux, sans homologues cellulaires, provoquent la formation
de tumeurs?
Il est connu que la plupart des tumeurs
sont le résultat d'une mutation dominante. C'est à dire que la cellule
acquiert une fonction qui la pousse à se diviser quand elle ne devrait
pas (Figure 17). Par exemple, les mutations dans un récepteur à activité
tyrosine kinase comme erb-B qui rendent l'activité tyrosine kinase
constitutive induiront une croissance cellulaire aberrante, même si une
seule des copies alléliques de cet oncogène cellulaire est touchée.
L'allèle muté est donc dominant sur l'allèle sauvage.
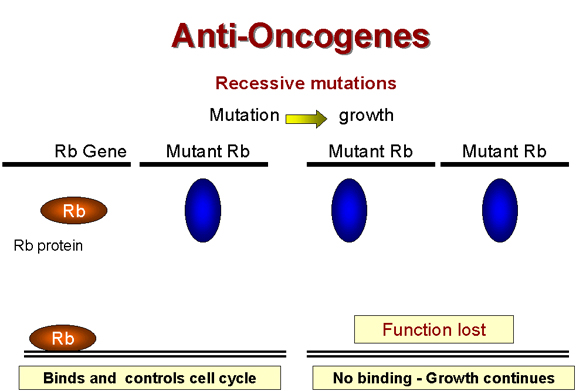
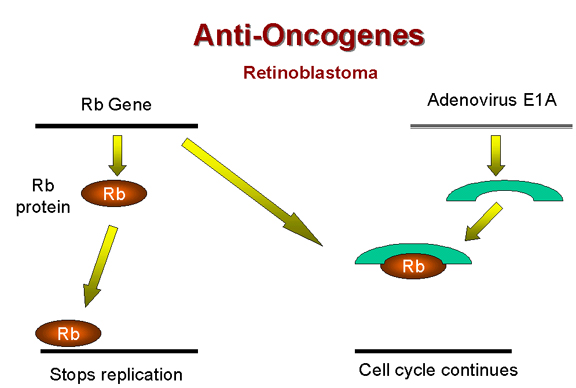
Le rétinoblastome – une
tumeur récessive
Il existe une classe de tumeurs qui ne
respectent pas la règle générale concernant la dominance des
mutations transformantes. Dans le cas du forme héréditaire du
rétinoblastome, la lésion génétique (tombant dans le gène RB1)
provoquant le cancer est récessive – ce qui indique qu'elle est
associée à une perte de fonction du gène. Des mutations "perte de
fonction" sont récessives car il faut des lésions génétiques dans
les deux copies alléliques du gène pour inactiver la fonction du
gène. Un organisme, ou une cellule, hétérozygote pour la mutation
possède toujours un phénotype normal.
Il semble donc que la fonction du gène
RB1 est de supprimer ou de limiter la division cellulaire. Le gène
RB1 est donc un gène suppresseur de tumeur, ou anti-oncogène. En
présence d'une mutation du gène RB1 à l'état homozygote, le produit
de gène RB1 sera totalement absent, et la cellule croîtra de façon
anormale, car le suppresseur de croissance n'est plus présent. En
revanche, à l'état hétérozygote, la copie non-muté du gène RB1 reste
fonctionnelle, et la croissance cellulaire peut être contrôlée
normalement. Le produit du gène RB1 a été identifié. Il s'agit d'une
protéine nucléaire d'un poids moléculaire de 105 kDa appelée
p105-Rb.
Précédemment, nous avons noté que le
gène précoce de l'adénovirus E1A, qui est nécessaire pour la
formation de tumeurs par ce virus, forme un complexe avec la
protéine p105-Rb (Figure 18). Il semble donc que l'activité
transformant de l'adénovirus est due à l'inactivation d'un anti-oncogène
cellulaire. Le même mécanisme est impliqué dans la formation de
tumeurs par le virus SV40, car l'antigène T également inactive la
protéine p105-RB.
La protéine p53 et le
cancer humain
La protéine p53 fut identifiée en 1979
chez les cellules de rongeur transformées par le virus SV40, et son
nom fut dérivé de l'observation qu'il s'agissait d'une protéine
cellulaire de 53 kDa associée à l'antigène T du virus SV40. Par la
suite, des mutations dans le gène TP53 (qui code pour la protéine
p53) ont été retrouvées dans de nombreux types de cancer. En effet,
les altérations, ou la perte totale, du gène TP53 semblent
impliquées dans le développement de la majorité des cancers humains.
Par exemple, l'on estime que le gène TP53 est touché chez 80% des
cancers du côlon, et même chez 60% des cancers (tous types confondus).
Initialement, on pensait que le produit
du gène TP53 agissait comme un oncogène, mais des recherches plus
approfondies ont montré le contraire; la protéine p53 est, comme la
p105-Rb, un suppresseur de tumeur. La protéine p53 a même été
désignée comme le "Gardien du Génome" car elle régule les multiples
composants du système de contrôle de dommage à l'ADN.
Quel est le fonctionnement de la
protéine p53 dans une cellule normale? A l'état de base, il y a
seulement quelques copies de la protéine p53 présentes dans une
cellule saine, et celles-ci sont constamment renouvelées, par une
dégradation constitutive de la protéine p53. Par contre, si une
cellule commence à se diviser après avoir subi un traitement qui
endommage l'ADN (par exemple, une exposition aux rayonnements UV, ou
un traitement par des mutagènes chimiques), la dégradation de la p53
cesse et le niveau de protéine p53 augmente. L'augmentation du
niveau de la p53 arrête la réplication de l'ADN, et bloque la
progression du cycle cellulaire.
La protéine p53 est un facteur de
transcription. Quand elle s'accumule, la p53 se fixe à des sites
spécifiques sur les chromosomes et induit l'expression d'autres
gènes qui eux, arrêtent la mitose. L'accumulation de la p53 peut
également déclencher l'apoptose. Le choix entre l'arrêt du cycle
cellulaire et l'induction de l'apoptose dépend de l'état
d'activation cellulaire ; par exemple, la présence de nombreuses
lésions d'ADN non réparées peut conduire à la production soutenue de
p53 qui condamne la cellule à la mort par apoptose.
Le syndrome Li-Fraumeni est une maladie
héréditaire rare provoquée par des lésions dans le gène TP53. Les
personnes atteintes ont un risque fortement accru du développement
de multiples types de cancer. Dans la plupart des cas, une mutation
ponctuelle résulte en l'expression d'une forme tronquée de la
protéine p53 qui est incapable de se lier à son site de fixation sur
l'ADN, et donc ne peut pas arrêter la réplication de l'ADN et le
cycle cellulaire. Comme pour le gène RB1 on pourrait s'attendre à ce
que les mutations dans TP53 soient récessives, car l'allèle sauvage
présent dans une cellule hétérozygote, ayant conservé sa fonction,
devrait être capable de bloquer la réplication de l'ADN. Néanmoins,
le syndrome Li-Fraumeni est une maladie génétique dominante – ce qui
indique que la fonction du p53 est inhibée même chez les cellules
(et chez les individus) hétérozygotes. Les mutations dans le gène
TP53 sont dominantes car la forme fonctionnelle de la protéine p53
est un homo-tétramère. Les tétramères qui incorporent une copie
mutée de p53 sont inactifs (même si les trois autres copies de p53
dans le tétramère sont normales), ce qui mène à un effet dominant-negatif
des mutations dans TP53.
Puisque les mutations héréditaires dans
TP53 sont très rares, il est clair que la majorité des mutations de
TP53 chez les cellules cancéreuses sont des mutations ponctuelles
somatiques induites par des facteurs environnementaux tels que les
rayonnements UV, les aflatoxines (produites par des moisissures sur
du maïs ou sur la pâte d'arachide), ou le benzopyrène dans la fumée
des cigarettes. Certaines de ces mutations dotent la protéine p53 de
nouvelles fonctions aberrantes, comme une résistance accrue à
l'apoptose, et ces mutations sont associées avec le développement de
tumeurs très agressives.
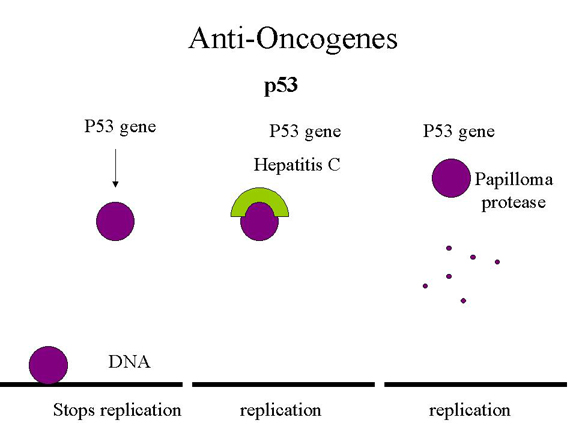
Quelle est la relation entre les
mutations dans TP53 et les virus transformants à ADN ? Comme pour la
protéine p105-Rb, les virus transformants à ADN doivent inactiver la
protéine p53 afin de permettre la réplication de leur ADN. Ainsi, le
virus SV40 (antigène T), les adénovirus (protéine E1B), et les
papillomavirus (protéine E6) expriment les protéines précoces qui se
fixent sur la protéine p53 et l'inactivent ou alors provoquent la
dégradation de la p53 (Figure 19).
|
| |
Résumé – Virus transformant,
oncogènes et anti-oncogènes
Chez les rétrovirus à fort potentiel transformant, le virus porte un
oncogène viral, qui est une forme mutée d'un oncogène cellulaire. La
présence de l'oncogène viral rend ce type de virus incapable de se
répliquer de façon autonome.
Chez les rétrovirus à faible potentiel transformant, l'intégration du
génome viral à proximité d'un oncogène cellulaire active l'expression de
ce dernier. Le virus ne porte pas d'oncogène viral – il est donc capable
de se répliquer de façon autonome.
Chez les virus transformant à ADN, un ou plusieurs gènes précoces du
virus inactivent les anti-oncogènes cellulaires p53 et p105-Rb.
L'inactivation de ces protéines cellulaires est essentielle pour le
virus, car cela permet la réplication de l'ADN viral. La carcinogénèse
est liée à l'intégration d'une partie du génome viral dans l'ADN
cellulaire, dans le cadre de l'infection d'une cellule non-permissive
pour le virus.
Par la suite, nous verrons à quel point ces trois mécanismes d'induction
de cancer par des virus dans des systèmes modèles sont pertinents pour
les cancers humains associés aux infections virales.
|
|
 Papilloma virus Copyright 1994 Veterinary Sciences
Division, Queens University Belfast
Papilloma virus Copyright 1994 Veterinary Sciences
Division, Queens University Belfast
 Papilloma virus © Dr Linda M.
Stannard 1995 (reproduit avec permission)
Papilloma virus © Dr Linda M.
Stannard 1995 (reproduit avec permission)
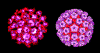 Papilloma virus. Image de microscopie électronique
colorisée par ordinateur. Tous les 72 capsomères sont des pentamères de
la protéine de capside majeurs. © Dr Linda M.
Stannard 1995 (reproduit avec permission)
Papilloma virus. Image de microscopie électronique
colorisée par ordinateur. Tous les 72 capsomères sont des pentamères de
la protéine de capside majeurs. © Dr Linda M.
Stannard 1995 (reproduit avec permission)
Figure 20
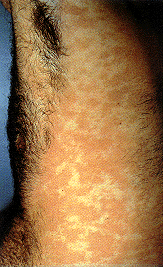
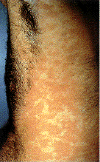 Epidermodysplasia verruciformis. Cette éruption
erythémateuse étendue, avec prurite était provoquée par une infection au
HPV. International Association of Physicians in
AIDS Care
Epidermodysplasia verruciformis. Cette éruption
erythémateuse étendue, avec prurite était provoquée par une infection au
HPV. International Association of Physicians in
AIDS Care

Epidermodysplasia verruciformis. Lésions verruciformes hypérkératosées sur
le dos de la main.

Epidermodysplasia verruciformis. Vue histopathologique: Koliocytes et
dysplasie modérée dans l'épiderme. (coloration Hématoxyline-Eosine
x100). D'après Reza Mahmoud Robati MD, Afsaneh Marefat MD, Marjan Saeedi
MD, Mohammad Rahmati-Roodsari MD, Zahra Asadi-Kani MD. Dermatology
Online Journal 15 (4): 8, 2009 (utilisé sous licence Creative Commons)
 Carcinome verruciforme. L'épithélium montre une maturation de surface,
parakératose et hypérkératose, en l'absence de cellules atypiques. On
distingue un léger infiltrat inflammatoire chronique.
D'après le Johns Hopkins Autopsy Resource (JHAR) Image Archive.
Carcinome verruciforme. L'épithélium montre une maturation de surface,
parakératose et hypérkératose, en l'absence de cellules atypiques. On
distingue un léger infiltrat inflammatoire chronique.
D'après le Johns Hopkins Autopsy Resource (JHAR) Image Archive.
Figure 21
 Figure 22
Figure 22
Verrues dans la région anale du périnée. Les verrues génitales (Condylomata
acuminata) sont des maladies sexuellement transmissibles provoquées
par le virus du papillome humain (HPV)
CDC
|
Virus provoquant des cancers humains
Papillomaviridae
Généralités
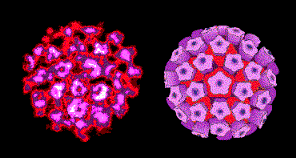
Les Papillomaviridae sont de petits virus à ADN icosaédriques non-enveloppés
(Figure 20). La protéine majeure de capside, VP1, est présente sous
forme de 72 pentamères dont chaque pentamère est associé à une molécule
d'une protéine de capside mineure, soit VP2 ou VP3. L'ADN est complexé
avec des protéines histones codées par la cellule hôte.
Les papillomavirus ont un génome en ADN
double brin circulaire d'environ 8 kilobases de longueur, codant pour 7
protéines précoces (E1 à E7) et les protéines tardives formant la
capside (VP1, VP2/3). Ils provoquent des verrues et des cancers chez
l'Homme ainsi que chez l'animal.
Il existe plus de 100 génotypes de virus du
papillome humain (HPV), dont la plupart ne sont pas associés aux
cancers. Ceci dit, il a été estimé que les différents HPV sont
responsables de 16% des cancers féminins dans le monde et de 10% de
l'ensemble des cancers.
Cancers associés
aux Papillomaviridae
Les HPV qui provoquent des verrues ne sont généralement pas associés aux
cancers, mais dans certains cas, des verrues peuvent se convertir en
carcinomes malins. Cela se produit, par exemple, chez les patients
atteints d'épidermodysplasie verruciforme.
L'épidermodysplasie verruciforme (Figure
21), également connu comme la dysplasie Lewandowsky-Lutz, est une
maladie très rare. Il s'agit d'une mutation autosomique récessive dans
les gènes EVER1 ou EVER2, qui permet une réplication anormale et
incontrôlée du virus du papillome. Cela se traduit par la croissance de
macules et papules squameuses sur de nombreuses parties du corps, mais
surtout sur les mains et les pieds. L'épidermodysplasie verruciforme,
qui est associée à un risque élevé de carcinome cutané, est généralement
associée aux HPV de type 5 et 8 (mais d'autres types peuvent également
être impliqués). Ces virus sont très répandus (infectant jusqu'à 80% de
la population) et provoquent des infections qui sont généralement
asymptomatiques chez le sujet sain.
Les papillomavirus sont également associés à des carcinomes du pénis, de
l'utérus, du col de l'utérus et de l'anus, et les verrues génitales
(Figure 22) peuvent parfois évoluer en carcinomes.
Les carcinomes du larynx, de l'œsophage et du poumon ressemblent
fortement au carcinome cervical sur le plan histologique, et un fort
lien de causalité entre certains cancers oro-pharyngés et le virus HPV16
a été démontré.
Les cancers vulvaires, ceux du pénis et du
col de l'utérus sont associés aux HPV de type 16 et 18 (et dans un
moindre degré HPV types 31, 33 et 45) mais les HPV génitaux les plus
courants sont les HPV de types 6 et 11. Comme on pouvait s'y attendre si
elles sont en effet les causes de certains cancers, les types 16 et 18
sont capables de transformer des kératinocytes humains en culture
cellulaire. Dans une étude allemande, il a été montré que 1 femme sur 30
infectées par le HPV de type 16 développera une maladie maligne tandis
que 1 personne sur 500 infectées développera un cancer du pénis ou de la
vulve. Étant donné que toutes les personnes infectées ne développent un
cancer, il y a probablement d'autres cofacteurs qui jouent un rôle
important dans l'évolution de la maladie. Les personnes infectées par le
VIH ou atteints d'un SIDA clinique ont un risque accru de cancers
associés au HPV comme les patients atteints d'autres formes
d'immunosuppression.
L'observation qu'un virus se trouve généralement en association avec une
maladie (souvent, dans le cas des tumeurs, la présence d'une copie du
génome viral dans les cellules néoplasiques) ne prouve pas que ce virus
a causé le cancer. Par exemple, l'hypothèse selon laquelle la présence
du virus dans les cellules tumorales serait une conséquence (plutôt que
la cause) de la transformation maligne, est également cohérente avec ce
type de données. Néanmoins, dans de nombreux cas, les données
épidémiologiques sont très convaincantes (par exemple, le HPV est
présent dès les premiers stades du développement du cancer, et donc sa
présence ne peut pas être une conséquence de la transformation maligne,
qui survient plus tardivement). De plus, l'efficacité du vaccin anti-HPV
dans la prévention du cancer du col de l'utérus constitue une
démonstration indéniable du rôle causal de l'infection virale dans le
développement de ce cancer.
Mécanismes moléculaires impliqués dans la
carcinogénèse
Comme les Polyomaviridae, les Papillomaviridae ont besoin de manipuler
le cycle cellulaire de la cellule hôte pour assurer la réplication de
l'ADN viral. Chez les HPV à fort potentiel transformant (notamment, les
HPV de type 16 et 18) les protéines précoces E6 et E7 possèdent des
fonctions analogues à celles de l'antigène T du virus SV40. La protéine
E6 interagit avec la p53, et induit sa dégradation par le protéasome,
tandis que la protéine E7 interagit avec plusieurs protéines cellulaires
impliquées dans la régulation du cycle cellulaire, y compris la p105-Rb.
L'expression continue des protéines E6 et
E7 lors d'une infection par HPV à fort potentiel transformant mène à une
dysplasie des cellules épithéliales infectées, caractérisée par des
lésions histologiques de bas grade, détectable par frotti cervical.
L'intégration de la région “précoce” du
génome du HPV dans l'ADN de la cellule hôte semble précéder la
progression des lésions dysplasiques de bas grade vers des néoplasies
malignes, et chez plus de 90% des carcinomes du col de l'utérus, l'ADN
viral est intégré dans l'ADN cellulaire. Puisque cette intégration est
un événement aléatoire qui ne fait pas partie du cycle de réplication
viral, elle ne survient pas systématiquement, et la majorité des lésions
de bas grade ne vont pas progresser vers un état cancéreux.
|
| |
|
 Herpesvirus en microscopie électronique à transmission. © Dr Linda M.
Stannard 1995 (reproduit avec permission)
Herpesvirus en microscopie électronique à transmission. © Dr Linda M.
Stannard 1995 (reproduit avec permission)
 Capside du virus Herpes Simplex, d'après F.P.Booy, W.W.Newcomb, B.L.Trus,
J.C.Brown, T.S.Baker, and A.C.Steven, in CELL, Vol 64 pp 1007-1015,
March 8, 1991
Capside du virus Herpes Simplex, d'après F.P.Booy, W.W.Newcomb, B.L.Trus,
J.C.Brown, T.S.Baker, and A.C.Steven, in CELL, Vol 64 pp 1007-1015,
March 8, 1991
 Virus Herpes Simplex en microscopie électronique à
transmission (x169 920) © Dennis Kunkel Microscopy
inc. (reproduit avec permission).
Virus Herpes Simplex en microscopie électronique à
transmission (x169 920) © Dennis Kunkel Microscopy
inc. (reproduit avec permission).
Figure 23
 Figure 24A
Figure 24A
Virus Epstein-Barr
 Figure 24B
Figure 24B
Lymphome de Burkitt provoqué par le virus Epstein-Barr (D'après le Johns
Hopkins Autopsy Resource (JHAR) Image Archive.)
 Figure 24C
Figure 24C
Distribution mondiale du lymphome de Burkitt.
A

B
 Figure 24D
Figure 24D
Frottis sanguins d'un individu sain (A) et d'un patient atteint d'une
mononucléose (B). © Gloria J. Delisle and Lewis Tomalty, Queens
University Kingston, Ontario, Canada and The Microbe Library
 Figure 24E
Figure 24E
Etape précoce de leucoplasie orale chevelue (OHL) sur le
bord latéral de la langue. L'infection par le VIH réduit la réactivité
immunologique, et l'environnement intra-orale est une cible de choix
pour les infections chroniques secondaires et les processus
inflammatoires, y compris la leucoplasie orale chevelue, qui est
provoquée par le virus d'Epstein-Barr dans des conditions
d'immunosuppression.
 Figure 24F
Figure 24F
Cytomegalovirus en microscopie électronique à
transmission (x49,200) CDC
|
Gammaherpesvirinae
Les Herpesviridae (Figure 23) sont beaucoup plus grands que les
Papillomaviridae et le Polyomaviridae, ayant des génomes de 100 à 200
kilobases de longueur. En raison de leur grande taille, il reste encore
beaucoup à découvrir au sujet de la façon dont ces virus transforment des
cellules.
Parmi les différents Herpesvirus qui infectent l'Homme, les virus
Epstein-Barr (EBV, ou HHV4 Figure 24A) et le virus associé au sarcome de
Kaposi (KSHV, ou HHV8) sont impliqués dans le développement de cancers
humains. Ces deux virus constituent la sous-famille des γ-Herpesvirinae, et
la capacité à induire des cancers est l'un des nombreuses ressemblances
entre ces deux virus.
Epstein-Barr virus (Human Herpesvirus 4 - HHV4)
Le HHV4 infecte principalement les lymphocytes B et les cellules
épithéliales. Comme tous les Herpesviridae, le HHV4 a la possibilité
d'alterner entre des cycles d'infection lytique, et des infections
latentes. L'infection des cellules épithéliales est généralement lytique,
et le virus est activement produit par des cellules épithéliales
infectées, tandis que l'infection des lymphocytes B mène à une infection
latente, caractérisée par l'expression d'un nombre limité des gènes
viraux.
L'infection par le HHV4 est extrêmement fréquente – aux Etats-Unis, plus
de 80% de la population est séropositive pour le HHV4 avant l'âge de 20
ans. Les infections lors de la jeune enfance sont le plus souvent
asymptomatiques, ou sous-cliniques, tandis que les infections chez
l'adolescent ou chez l'adulte peuvent provoquer la mononucléose
infectieuse, une infection des lymphocytes B qui provoque une
prolifération bénigne de ceux-ci (Figure 24D), qui se résout
spontanément chez des individus immunocompétents. Dans le cas d'une
immunosuppression (par exemple, chez les personnes atteintes du SIDA),
l'infection par le HHV4 peut provoquer la leucoplasie chevelue buccale
("Oral Hairy Leukoplakia", ou OHL - Figure 24E). Chez des individus
porteurs d'une lésion génétique rare, l'infection par le HHV4 provoque
la maladie lymphoproliferative liée au chromosome X, (aussi connu comme
"Maladie de Duncan"), caractérisée par une hyperactivation du système
immunitaire, liée à l'incapacité de contrôler l'infection par le HHV4.
Néanmoins, l'infection par le HHV4 est associée à plusieurs cancers:
Le cancer du nasopharynx, particulièrement en Chine et en Asie du
sud-est. Un cofacteur alimentaire semble être responsable de la
distribution géographique de ce cancer.
Le lymphome de Burkitt (Figure 24B), qui est un lymphome pédiatrique de
cellules B observé aux zones tropicales, et en particulier dans les
zones où le paludisme est endémique (Figure 24C).
Le lymphome de Hodgkin (encore un lymphome de cellules B). L'ADN viral
est présent dans les cellules malignes chez environ 40% des patients
touchés.
Des lymphomes B non-Hodgkiniens chez les patients immunodéprimés –
notamment chez les receveurs de greffe d'organe, ou chez les patients
atteints du SIDA.
L'un des objectifs des études sur le HHV4, est de comprendre pourquoi ce
virus provoque des maladies bénignes chez la grande majorité des
personnes infectées, mais peut causer des cancers chez d'autres.
Mécanismes moléculaires de carcinogénèse
Le HHV4 provoque des lymphomes chez le marmoset (un membre des
Platyrrhini, les singes du nouveau monde), et il est capable de
transformer des lymphocytes B humains in vitro. Dans les lymphocytes B
infectés, un nombre restreint de gènes viraux (codant pour 9 protéines
de latence, et deux ARNs non-codant) sont exprimés, et le génome viral
est maintenu sous forme d'épisome (c'est à dire, de l'ADN double-brin
circulaire, non-intégré dans le génome de la cellule hôte). Les
protéines de latence induisent la prolifération de la cellule infectée.
En particulier, la protéine LMP1, insérée dans la membrane plasmique de
la cellule infectée, agit comme un oncogène viral, car elle active de
multiples voies de signalisation intracellulaire qui induisent la
prolifération, et inhibent l'apoptose (c'est à dire, la mort cellulaire
programmée) de la cellule infectée.
La prolifération des lymphocytes B infectés de façon latente par le HHV4
est directement responsable de la mononucléose. Chez les personnes
immunocompétentes, la présence de protéines virale dans les lymphocytes
B infectées permet leur élimination par le système immunitaire, et la
disparition des symptômes. Néanmoins, d'autres lymphocytes B infectés,
qui expriment une seule protéine virale (l'antigène nucléaire EBNA-1)
persistent, et l'infection latente est maintenue tout le long de la vie
de la personne infectée.
Le développement d'une prolifération maligne des lymphocytes B suite à
une infection par le HHV4 nécessite la présence d'une ou de plusieurs
cofacteurs, qui sont:
L'immunosuppression
Dans ce cas, les lymphocytes B exprimant les protéines de latence du
virus ne peuvent pas être éliminés par la réponse immune, et la
prolifération bénigne de ces cellules peut basculer vers une
prolifération maligne. Le résultat est le développement d'un lymphome B
chez les sujets immunodéprimés.
L'activation de l'oncogène cellulaire c-myc par translocation
Les cellules malignes du lymphome de Burkitt sont caractérisées par la
présence d'une translocation chromosomique entre le chromosome 8 et le
chromosome 14, qui met le gène c-myc à proximité du gène codant pour la
chaîne lourde des immunoglobulines. Le gène c-myc est donc exprimé de
façon constitutive, ce qui engendre la prolifération cellulaire, même en
l'absence de l'expression de l'oncogène viral LMP1.
Il a été proposé que l'hyperstimulation de la réponse immunitaire
provoquée par des infections à répétition au Plasmodium falciparum (le
parasite responsable du paludisme) augmente la probabilité de la
survenue de la translocation 8:14, ce qui expliquerait l’association
épidémiologique entre l'incidence du lymphome de Burkitt et le paludisme.
Il a aussi été observé que l'aire de répartition de la plante Euphorbia
tirucalli correspond aux zones d'Afrique où la prévalence du lymphome de
Burkitt est élevée. Des produits naturels présents dans la sève de la
plante sont capables d'induire des translocations chromosomiques, y
compris les translocations du chromosome 8 entraînant l'activation de
l'oncogène c-myc. Dans ce cas, l'exposition à un carcinogène chimique
produit par l'E.tirucalli serait un cofacteur important dans l'étiologie
du lymphome de Burkitt.
|
| |
Kaposi Sarcoma Herpes Virus (Human herpes
virus 8)
Le HHV8 infecte des lymphocytes et les cellules épithéliales et
endothéliales. Il est l'agent causal du sarcome de Kaposi, qui est
caractérisé par une hyperplasie des cellules endothéliales. Il est
également associé à un certain nombre de cancers hématologiques, y
compris la maladie de Castleman (parfois mal-épelé "Castelman")
multifocale, et le lymphome primitif des séreuses (un lymphome
diffus à grandes cellules B). Tous les cancers provoqués par le HHV8
sont des cancers relativement rares.
Le HHV4 et le HHV8 sont aussi associées à des lésions et les tumeurs
buccales chez les patients infectés par le VIH. Parmi ces maladies,
la leucoplasie chevelue buccale est une maladie bénigne qui provoque
des épaississements blancs sur l'épithélium de la langue contenant
des foci de réplication virale (Figure 24E).
En Amérique du nord, l'Europe du nord et en Asie, la séroprévalence
du HHV8 est inférieur à 5%. Elle est supérieure à 50% dans l'Afrique
sous-saharienne, et environ 30% dans le bassin méditerranéen. Même
dans les zones endémiques pour l'infection par le HHV8, l'incidence
des cancers associés à ce virus est faible, ce qui implique que
l'immense majorité des infections à HHV8 ne résultent pas en un
cancer. Par exemple, en l'absence d'infection par le VIH, seul
1/5000 à 1/20000 des personnes séropositives pour la HHV8 vont
développer un sarcome de Kaposi.
Mécanismes moléculaires de carcinogénèse
Le génome du HHV8 code pour plusieurs oncogènes viraux impliqués
dans la transformation des cellules infectées. Notamment, le gène
LANA-1 interagit avec les protéines cellulaires p53 et p105Rb et
modifie la régulation du cycle cellulaire chez les cellules
infectées. La protéine LANA-1 du HHV8 joue donc un rôle semblable à
celui de l'antigène T du virus SV40. D'autres oncogènes viraux du
HHV8 induisent la prolifération cellulaires, et inhibent la mort
apoptotique des cellules infectées.
Comme pour le HHV4, l'immunosuppression est un cofacteur important
pour le développement de cancers liés au HHV8. Par exemple, en
l'absence d'une co-infection par le VIH, il a été estimé que
seulement 1 sur 5000 à 1 sur 20 000 des personnes infectées par le
HHV8 vont développer un sarcome de Kaposi. Chez les personnes
infectés par le VIH et le HHV8, le risque de développer un sarcome
de Kaposi augmente avec la progression de l'infection par le VIH –
plus le taux de lymphocytes T CD4+ est bas, plus le risque de
développement de sarcome de Kaposi est élevé.
|
|
 Lymphocyte T humain infecté par le HTLV-1. Le virus se trouve en amas en
haut à droite (coloration mauve clair). © Dennis Kunkel Microscopy, Inc.
(reproduit avec permission)
Lymphocyte T humain infecté par le HTLV-1. Le virus se trouve en amas en
haut à droite (coloration mauve clair). © Dennis Kunkel Microscopy, Inc.
(reproduit avec permission)
Figure 25 |
Retroviridae
Deux rétrovirus humains, le HTLV-1et le HTLV-2, sont associés avec le
développement de cancers.
HTLV-1
Le HTLV-1 (Figure 25) a été isolé en 1980 par l'équipe de R. Gallo à
partir d'un prélèvement d'un patient afro-américaine. Il est associé avec la
leucémie T de l'adulte (ATL). Les régions avec la plus forte prévalence de
l'infection sont les îles sud-ouest de l'archipel japonais (12 à 16% de la
population infectée), les caraïbes (2 à 6% de la population infectée),
l'Amérique latine, l'Afrique équatoriale, le nord-est de l'Iran et le
Mélanésie. De plus, l'infection par le HTLV-1 est observée à un taux
beaucoup plus élevé que celle de la population générale chez les peuples
autochtones australiens et des Inuits du Canada.
Dans toutes les régions touchées par le virus, la séroprévalence augmente
avec l'âge, en particulier chez les jeunes femmes.
Au niveau mondial, on estime que 15 à 20 millions de personnes sont
infectées par le HTLV-1, et 2 à 10% de ces personnes vont développer une
maladie associée à ce virus.
Dans la région des Caraïbes, le virus est associé à un neuromyélopathie
connu sous le nom paraparésie spastique tropicale (TSP) et aussi avec la
dermatite infectieuse. Au Japon, le HTLV-1 provoque une maladie semblable à
la TSP appelée "myélopathie associée au HTLV" (HAM). Au Japon, le HTLV -1
est également associée à une uvéite. D'autres maladies associées au virus
sont l'arthrite et la polymyosite.
La TSP est une maladie qui affecte la matière blanche et grise de la moelle
épinière (une myélopathie). Il s'agit d'une maladie inflammatoire chronique
et progressive qui provoque une spasticité progressive des jambes,
l'incontinence et la constipation. Environ 1 à 4 % des patients infectés par
le HTLV-1 vont développer une TSP.
Le HTLV-1 se transmet par trois voies principales:
i. Transmission mère à l'enfant via l'allaitement. Environ 15 à 20%
des enfants nourris par le lait maternel des femmes séropositives pour
le HTLV-1 sont infectés. Ces enfants deviennent des porteurs chroniques
du virus.
ii. Sexuelle: La transmission homme-femme est plus fréquent
qu'inversement. Puisque le nombre de partenaires sexuels augmente avec
l'âge, la transmission sexuelle semble expliquer l'augmentation du taux
de séropositivité avec l'âge observé chez les femmes. La transmission
homme-homme lors des rapports sexuels est également observée.
iii. Des produits sanguins contaminés (transfusions sanguines)
|
| |
Mécanismes moléculaires de
carcinogénèse
L'ATL est un cancer qui se déclare au moins 20
à 30 ans après l'infection initiale par le HTLV-1. Il est généralement la
conséquence d'une infection dès l'enfance - le développement d'un ATL suite
à une infection à l'âge adulte est possible, mais il est rare. Le risque
cumulé de développer un ATL à un moment dans sa vie chez les porteurs du
HTLV-1 est de 6 à 7% chez les hommes, et de 2 à 3% chez les femmes. Le
HTLV-1 n'est donc pas un rétrovirus à fort potentiel transformant, car la
pénétrance est faible (c'est à dire que >90% des porteurs chroniques du
HTLV-1 ne vont jamais développer un ATL), et le processus de carcinogénèse
dure plusieurs décennies. L'absence d'un v-onc classique dans le génome du
HTLV-1 est cohérente avec ces données épidémiologiques.
L'ADN proviral du HTLV-1 se retrouve intégré dans le génome des cellules
leucémiques de l'ATL. Cependant, cette insertion ne semble pas activer un
oncogène cellulaire, car les sites d'insertion de l'ADN proviral du HTLV ne
sont pas à proximité d'un gène cellulaire en particulier. Le mécanisme de
carcinogénèse n'est donc pas le même que chez les rétrovirus à faible
potentiel transformant dans des systèmes modèles (comme par exemple le ALV).
La plupart des gènes viraux (gag, pol et env)
ne sont pas exprimés chez les cellules leucémiques de l'ATL. Par contre les
gènes accessoires du virus, tax et surtout HBZ, sont exprimés chez les
cellules leucémiques. Il semble donc que ces deux gènes viraux puissent agir
comme des v-onc, en particulier dans le cadre d'une infection abortive d'un
lymphocyte T. Le rôle, et même l'existence, du gène HBZ furent longtemps
ignorés, car ce gène est codé sur le brin anti-sens de l'ADN proviral tandis
que tous les autres gènes – gag, pol, env, et les gènes de régulation sont
codés sur le brin sens.
|
 Particules du virus de l'hépatite B, montrant deux cores
exposés (flèches)
Particules du virus de l'hépatite B, montrant deux cores
exposés (flèches)
 Particules du virus
de l'hépatite B
Particules du virus
de l'hépatite B
 Représentation schématique des particules infectieuses du
VHB (plus grandes - appelées particules de Dane), et des particules non-infectieuses
(plus petites) composées uniquement de la antigène S du VHB.
Représentation schématique des particules infectieuses du
VHB (plus grandes - appelées particules de Dane), et des particules non-infectieuses
(plus petites) composées uniquement de la antigène S du VHB.
Particules du virus de l'hépatite B
Les quatre images © Dr Linda M. Stannard 1995 (reproduit avec
permission)
Figure 26
 Cette femme est infectée par le virus de l'hépatite B et souffre d'un
cancer du foie. Elle était un ressortissant cambodgien et mourut 4 mois
après son arrivée dans un camp de réfugiés (l'espérance de vie moyenne
après le diagnostic de cancer du foie est de 6 mois)
Cette femme est infectée par le virus de l'hépatite B et souffre d'un
cancer du foie. Elle était un ressortissant cambodgien et mourut 4 mois
après son arrivée dans un camp de réfugiés (l'espérance de vie moyenne
après le diagnostic de cancer du foie est de 6 mois)
Coalition d'action pour l'immunisation (avec la
permission de Patricia Walker, MD, Clinique Ramsey Associates, St. Paul,
MN)
Figure 27 |
Les virus des hépatites chroniques
Le virus de l'hépatite B et
le carcinome hépatocellulaire
Le virus de l'hépatite B (VHB, Figure 26), étant un virus de la
classe VII de Baltimore, est un virus à rétrotranscription, et sa
mode de réplication ressemble plus à celui des rétrovirus qu'à celui
des virus transformant à ADN. Le virion renferme un génome en ADN
circulaire, qui est transcrit en ARN dans la cellule infectée pour
fournir non seulement des ARNm pour la production des protéines
virales, mais également des copies du génome en ARN qui seront
retranscrites en ADN par la transcriptase inverse du virus.
La biologie moléculaire et les pathologies liées au VHB sont
décrites dans le chapitre 18 et la deuxième partie du chapitre 19.
Le VHB représente un immense problème de santé mondiale, car il
fait partie des causes du carcinome hépatocellulaire (CHC, Figure
27), qui est l'un des cancers les plus fréquents au monde. Il existe
une très forte corrélation entre l'antigenémie pour la glycoprotéine
de surface du VHB (HbsAg) chez les porteurs chroniques du virus et
l'incidence du CHC. Par exemple, au Taiwan le risque de développer
un CHC est 217 fois plus élevé chez un porteur du VHB HbsAg+ que
chez un non-porteur, et la cirrhose du foie et le CHC sont
responsables de 51% des morts chez les individus infectés HbsAg+,
tandis que ces pathologies touchent seulement 2% de la population
générale.
Le virus de l'hépatite C et
le carcinome hépatocellulaire
Le virus de l'hépatite C (VHC) faisant partie de la famille
Flaviviridae est un virus ARN brin (+) avec une capside icosaédrique,
enveloppée. Il est responsable des hépatites jadis appelés "non-A,
non-B", et l'on estime qu'il y a plus de 200 millions porteurs de ce
virus au niveau mondial. Bien que les cas d'hépatite aigüe mortels
soient rares lors de la primo-infection, le VHC (comme le VHB) peut
provoquer une hépatite chronique. L'infection chronique provoque une
fibrose progressive du foie qui peut se développer en cirrhose et
ensuite en CHC. La progression de la pathologie étant lente – la
cirrhose et le CHC se déclarent jusqu'à 30 ans après l'infection
initiale - seule une minorité (environ 5%) des patients atteints par
une hépatite C chronique vont développer un CHC.
Mécanismes de carcinogénèse
par les VHB et VHC
Les hépatites virales chroniques sont les causes principales du
CHC, et l'on pense que ces virus sont responsables d'environ 70% des
cas de ce cancer. D'autres facteurs de risque, comme la consommation
d'alcool, sont également importants. Les mécanismes impliqués dans
la formation des tumeurs semblent très complexes, et dans une
cellule infectée par le VHB ou le VHC, les événements au niveau
moléculaire qui rendent cette cellule cancéreuse ne sont pas encore
bien établis. Néanmoins, les données indiquent que les protéines
virales contribuent directement à la transformation. L'antigène "x"
du VHB (Hbx), et la protéine du core et les protéines non-structurales
du VHC semblent interagir avec une variété de protéines cellulaires,
y compris les protéines p53 et p105-Rb. Cependant, ces observations
ne constituent pas de preuve définitive que ces interactions
moléculaires sont les causes de la transformation maligne.
La façon dont le VHB provoque des tumeurs est complexe; mais
l'intégration du génome viral dans l'ADN de la cellule hôte se
produit à un stade précoce de l'infection. L'expression des
protéines virales HBx et l'antigène HBS modifie le contrôle de la
réplication de l'ADN de l'hôte, et mène à la prolifération de la
cellule – ces gènes peuvent donc être considérés comme des oncogènes
viraux. Le gène suppresseur de tumeur TP53 est fréquemment inactivé,
soit par la présence de mutations somatiques chez les cellules
tumorales, soit par la fixation de la protéine HBx sur la protéine
p53. Cependant, de multiples voies de signalisation intracellulaire
sont modulées par les protéines du VHB dans le cadre du CHC, et le
processus de carcinogénèse ne se résume pas à simplement
l'inactivation de la protéine p53. L'expression des oncogènes viraux
peuvent également augmenter la sensibilité des hépatocytes aux co-carcinogènes,
tels que les aflatoxines, ou l'alcool.
Etant un virus à ARN de la famille des Flaviviridae (et non pas
un rétrovirus), le matériel génétique du VHC n'est jamais transcrit
en ADN, et ne peut donc pas s'intégrer dans le génome de la cellule
hôte. Néanmoins, la protéine virale non-structurale NS5A, et la
protéine C du virus interagissent avec de nombreuses protéines
cellulaires, y compris la protéine p53. Les protéines virales ont
donc la capacité d'inactiver des anti-oncogènes cellulaires.
L'initiation du CHC par le VHB semble se produire pendant les
premières années de la vie, car la plupart des patients touchés par
un CHC associé au VHB sont victimes d'une infection lors de
l'enfance. En revanche, dans le cas du VHC, le CHC se déclare
souvent à la suite d'une infection à l'âge adulte. Malgré une
différence importante au niveau de la biologie moléculaire de ces
deux virus – le VHB s'intègre dans le génome de la cellule hôte,
tandis que le VHC ne le fait pas – le processus de carcinogénèse
provoqué par ces deux virus peut être semblable. D'une part, les
mutations dans le gène TP53 sont fréquentes chez les CHC induits par
le VHB et par le VHC, et en l'absence de mutations de TP53, les
protéines virales sont capables d'inactiver la protéine p53 normale.
D'autre part, les deux maladies sont caractérisées par une
inflammation chronique du foie, qui est un facteur important dans le
développement de tumeurs.
|
|
|
 Retour à la section de virologie de Microbiologie et Immunologie On-line
Retour à la section de virologie de Microbiologie et Immunologie On-line
This page last changed on
Wednesday, February 19, 2014
Page maintained by
Richard Hunt
|
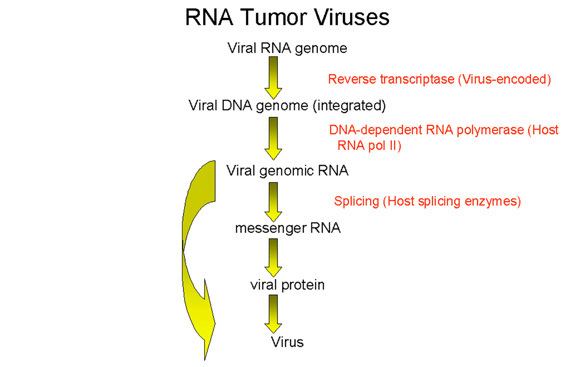 Figure 1
Figure 1 Figure 2A
Figure 2A Structure génomique d'un rétrovirus typique et d'un rétrovirus porteur
d'un oncogène viral (Virus de Sarcome de Rous)
Structure génomique d'un rétrovirus typique et d'un rétrovirus porteur
d'un oncogène viral (Virus de Sarcome de Rous) Oncogénèse par insertion du promoteur. La LTR agit comme promoteur,
induisant une expression abérrante d'un oncogène cellulaire.
Oncogénèse par insertion du promoteur. La LTR agit comme promoteur,
induisant une expression abérrante d'un oncogène cellulaire.  Le flux d'information chez un virus à ADN est semblable à
celui chez une cellule eucaryote.
Le flux d'information chez un virus à ADN est semblable à
celui chez une cellule eucaryote.  Adenovirus © Dr Stephen Fuller 1998
Adenovirus © Dr Stephen Fuller 1998 Adenovirus
en microscopie électronique à transmission. © Dr Linda M. Stannard 1995
(reproduit avec permission)
Adenovirus
en microscopie électronique à transmission. © Dr Linda M. Stannard 1995
(reproduit avec permission) Figure 24B
Figure 24B 
 Figure 24D
Figure 24D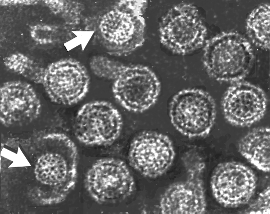 Particules du virus de l'hépatite B, montrant deux cores
exposés (flèches)
Particules du virus de l'hépatite B, montrant deux cores
exposés (flèches)