VIROLOGY - LECTURE SEVEN

HIV and AIDS lecture notes from Dr Richard Hunt
University of South Carolina School of Medicine
Department of Microbiology and Immunology
AN INDEX TO THESE PAGES MAY BE FOUND HERE

 From Dr. Milan V.Nermut of the National Institute
for Biological Standards and Control. Herts, U.K. Computer graphics by A.Davies
From Dr. Milan V.Nermut of the National Institute
for Biological Standards and Control. Herts, U.K. Computer graphics by A.Davies
WORLD WIDE WEB LINKS ARE TO BE FOUND IN VARIOUS SECTIONS OF THIS PAGE. MOST OF THESE LINKS COME FROM UNISCI OR MEDSCAPE. IN ADDITION, GO TO THIS PAGE FOR FURTHER HIV/AIDS INFORMATION INCLUDING
HIV NEWS -- RECENT UPDATES AND NEWS STORIES
HIV NEWS -- INFORMATION AND SUMMARIES FROM CONFERENCES ON HIV/AIDS AND ANTI-MICROBIAL AGENTS AND CHEMOTHERAPY
HIV NEWS FROM CDC
GENERAL WWW LINKS FOR HIV AND AIDS INFORMATION
CDC HIV/AIDS REPORTS 1997,1998,1999
FREQUENTLY ASKED QUESTIONS ABOUT HIV/AIDS
CURRENT STATUS OF SPECIFIC ANTI-HIV DRUGS AND LINKS TO DRUG DATA SHEETS
CURRENT STATUS OF ANTI-HIV VACCINES
How many people are living with HIV infection today?
Worldwide? Go hereHow many people are living with HIV infection in various regions of the world? Go here What proportion of infected people in various regions are women? Go here
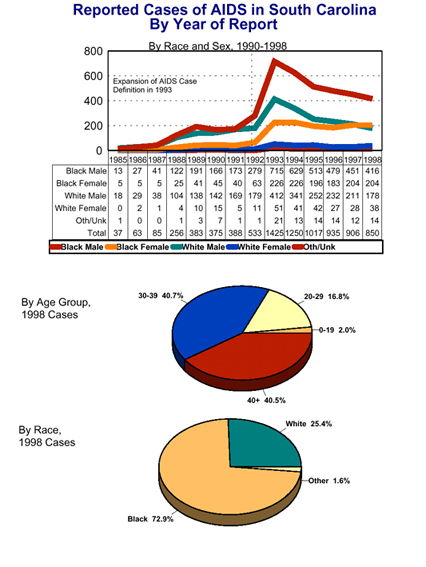
What are the major mechanisms of HIV transmission in different regions of the world? Go here What is the proportion of the adult population (15-49 years of age) in different regions of the world that is living with HIV or AIDS? Go here How many people have been infected by HIV in South Carolina? Go here or click on image
South Carolina HIV/AIDS data report from the Department of Health and Environmental Control - Go here - Requires Acrobat
Click on thumbnails
|
|
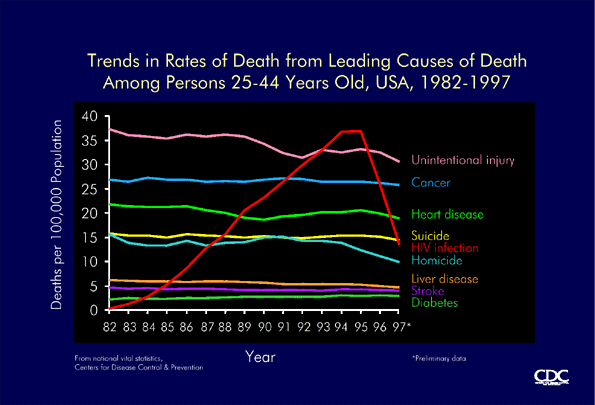
Death rates from leading causes of death in persons aged 25-44 years, USA, 1982-1997 (CDC) |
|
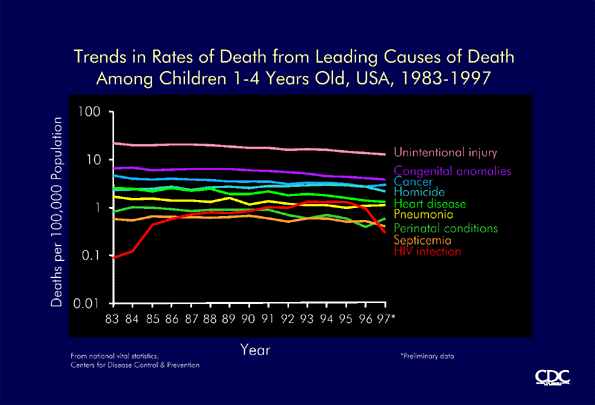
Trends in rates of death from leading causes of death in children 1-4 years old, USA, 1983-1997 (CDC) |
|
|
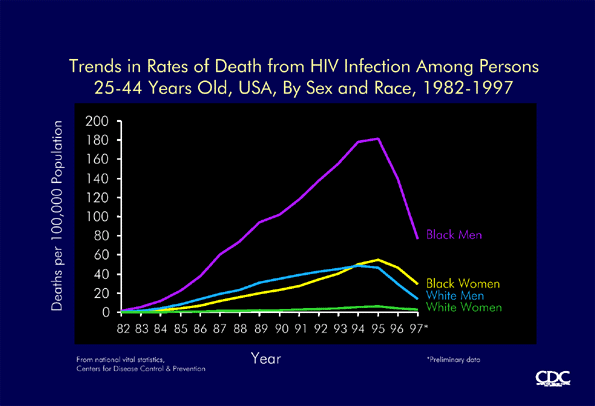
Trends in death rates from HIV infection among persons aged 25-44, USA, 1982-1997 by sex and race (CDC) |
|
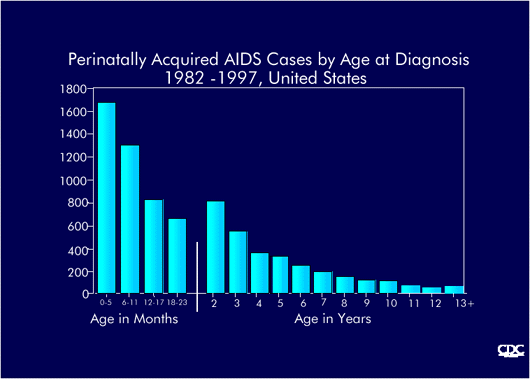
Perinatally-acquired AIDS cases by age at diagosis, USA, 1983-1997 (CDC) |
|
|
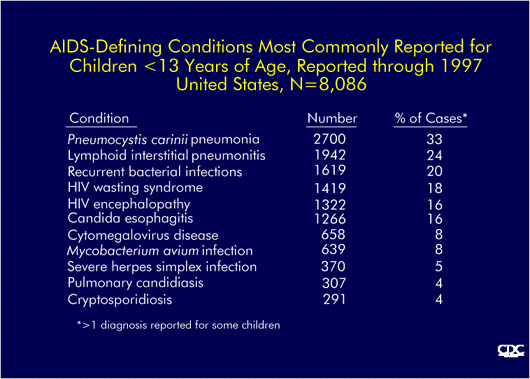
AIDS-defining conditions most commonly reported in children (CDC) |
|
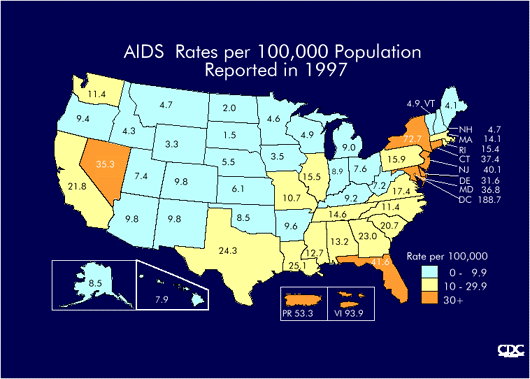
Incidence in USA |
Human immunodeficiency virus (HIV) is found in all cases of AIDS (acquired immunodeficiency disease syndrome). In the infected patient, it can be detected by the presence of anti-HIV antibodies or by the presence of the virus itself using polymerase chain reaction. The latter is very sensitive and can show HIV in situations in which it is not detectable immunologically.
HIV is a lentivirus, a class of retroviruses, and integrates into the genome in the same manner as other retroviruses. Unlike other retroviruses, which typically bud from the infected cell for a long period of time, HIV can lyze the cell or lie dormant for many years, especially in resting T4 lymphocytes; later, a recrudescence of viral production occurs that ultimately destroys the cell. It should be noted that while HIV may disappear from the cells of the circulation, viral replication and budding continues to occur in other tissues. In contrast to T4 lymphocytes, other HIV-infected cells do not show a pronounced cytopathic effect and may exhibit latency or continual budding. Unlike many other retroviruses, no copies of primate lentiviruses are found in the genome of target species and the virus not transmitted through the germ line.
![]() Progression of AIDS in the United States from 1979. Click on icon
at left to see moving .gif file CDC
Progression of AIDS in the United States from 1979. Click on icon
at left to see moving .gif file CDC
In 1981, clusters of cases of Kaposi's sarcoma were observed in patients in San Francisco and New York. Kaposi's sarcoma normally occurs in elderly Jewish men but these patients were all were young male homosexuals. Other diseases associated with immuno-compromisation also occurred in this same population; particularly of note were the occurrence of Pneumocystis pneumonia (which is an opportunistic infection) and lymphadenopathy. Pneumocystis pneumonia, which is caused by an intracellular parasite (Pneumocystis carinii), was also a very rare disease. Between 1967 and 1978 only 5 cases occurred in Los Angeles but five cases occurred in 1981 alone. Again, all patients were young male homosexuals. The number of sex partners of these patients appeared to be important in that the disease was particularly prevalent among promiscuous individuals and the partners of those individuals. Later, a similar immunodeficiency was found in intra-venous drug users who shared needles, persons who received blood transfusions and hemophiliacs. Moreover, the sex partners of these patients also got the disease. In view of this, it was obvious that an infectious agent was involved; this agent was either passed during sexual intercourse or by receiving blood from another person. The cell picture - the selective loss of T4 helper (CD4+) cells - suggests a virus. But the causative agent was difficult to identify at first because it does not grow on resting T4 cells. The discovery of HIV depended on the ability to grow the virus in vitro.
The disease was originally termed Gay-Related Immune Deficiency (GRID) but we now know it as Acquired Immuno-Deficiency Syndrome (AIDS). AIDS is invariably fatal (unless chemotherapeutic intervention occurs). The epidemic has resulted in the deaths of more than half of AIDS patients.

Lesions on the stomach of a patient with Kaposi's sarcoma (Click to enlarge) |
 The perivenular infiltrate of Kaposi sarcoma shows a mixture of spindle cells, inflammatory cells, and ectatic vascular spaces.
The Johns Hopkins Autopsy Resource
(JHAR). Image Archive.
The perivenular infiltrate of Kaposi sarcoma shows a mixture of spindle cells, inflammatory cells, and ectatic vascular spaces.
The Johns Hopkins Autopsy Resource
(JHAR). Image Archive. |
 Kaposi's sarcoma (skin). Bristol Biomedical Image Archive,
University of Bristol. Used with permission
Kaposi's sarcoma (skin). Bristol Biomedical Image Archive,
University of Bristol. Used with permission |
|
The clue to growing the virus came with the realization that, while it did not grow on resting cells, it would grow on T4 cells that had been activated with a cytokine called interleukin-1. The virus was finally isolated by Luc Montanier (Pasteur Institute, Paris) and Robert Gallo (NIH, Bethesda, USA). Montanier called the virus lymphadenopathy virus (LAV) and Gallo, who had discovered the first human leukemia virus (HTLV-1), named the virus HTLV-3. Today we know it as human immunodeficiency virus (HIV).
A similar cellular picture is seen in some cases of feline leukemia and HTLV-1, i.e. a selective
loss of a specific class of cells giving rise to immune suppression, further
suggesting that the cause of AIDS is a virus.
 Electron
micrograph of HIV
(click to enlarge) - Cone-shaped cores are sectioned in various orientations. Viral genomic RNA is located in the electron-dense wide end of core. CDC/Dr. Edwin P. Ewing, Jr.
epe1@cdc.gov
Electron
micrograph of HIV
(click to enlarge) - Cone-shaped cores are sectioned in various orientations. Viral genomic RNA is located in the electron-dense wide end of core. CDC/Dr. Edwin P. Ewing, Jr.
epe1@cdc.gov

 Click on thumbnail to enlarge
Click on thumbnail to enlarge
From the original infection, there is usually a period of 8-10 years before the clinical manifestations of AIDS occur; however, this period may be two years or less. Approximately 10% of patients succumb to AIDS within 2 to 3 years
a) Acute infection (acute retroviral syndrome)
Initially, HIV infection produces a mild disease that is self-limiting. This is not seen in all patients. In the period immediately after infection, virus titer rises (about 4 to 11 days after infection) and and continues at a high level over a period of a few weeks. The patient experiences some mononucleosis-like symptoms (fever, rash, swollen lymph glands (Information Box) but none of these are life-threatening. The result is an initial fall in the number of CD4+ cells and a rise in CD8+ cells but the numbers quickly return to near normal.
 HIV-1 budding from cultured lymphocyte. Transmission electron micrograph.
CDC/Dr. Edwin P. Ewing, Jr. epe1@cdc.gov
HIV-1 budding from cultured lymphocyte. Transmission electron micrograph.
CDC/Dr. Edwin P. Ewing, Jr. epe1@cdc.gov |
 Scanning electron micrograph of HIV-1 budding from cultured lymphocyte. Multiple round bumps on cell surface represent sites of assembly and budding of
virions (CDC)
Scanning electron micrograph of HIV-1 budding from cultured lymphocyte. Multiple round bumps on cell surface represent sites of assembly and budding of
virions (CDC) |
 Facial sarcoidosis in AIDS Bristol Biomedical Image Archive,
University of Bristol. Used with permission
Facial sarcoidosis in AIDS Bristol Biomedical Image Archive,
University of Bristol. Used with permission |
b) A strong cell-mediated and humoral anti-HIV immune defense
Cytotoxic B and T lymphocytes mount a strong
defense and virus largely disappears from circulation. During this period, more
than 10 billion new HIV particles are produced each day but they are rapidly cleared
by the immune system and
have a half life of only 5-6 hours (some estimates show a half life of minutes). There can be
up to 104 to 107 virus particles per ml of blood. (Titers of
infectious virus are much lower indicating that much of the plasma virus is defective or
neutralized). Most of this virus is coming from recently infected proliferating CD4+
cells. The infected cells that are producing this virus are destroyed either by the immune
system or by the virus (half life about 1 day). However, the rate of production of CD4+
cells can compensate for the loss of cells and a steady state is set up in which a very
small fraction of the resting memory CD4+ cells carry an integrated HIV genome.
Most CD4 cells at this stage are uninfected.
The virus disseminates to other regions including to lymphoid and nervous
tissue. This is the most infectious phase of the disease. Seroconversion occurs
between one and four weeks after infection.
c) A latent reservoir. As a result of the strong immune defense, the number of viral particles in the blood stream declines and the patient enters clinical latency. Little virus can now be found in the bloodstream or in peripheral blood lymphocytes. Initially, the number of blood CD4+ cells is only slightly decreased. Nevertheless, the virus persists elsewhere, particularly in follicular dendritic cells in lymph nodes and here viral replication continues. Virus can become trapped in the follicular dendritic cell network of lymphoid tissue. The virus is also replicated by macrophages. A small fraction of the productively infected CD4+ cells may survive long enough to revert back to the resting memory state (as do non-infected CD4+ memory cells). The resting memory cells do not express viral antigens but do carry a copy of the HIV genome which remains latent until the cells are reactivated by antigen. These memory cells have a great potential for stability and constitute a reservoir that may be very important in drug-based therapy.
Note: Although the number of HIV particles in the bloodstream falls during clinical latency, the virus is detectable. After the initial peak of virus, the virus reaches a "set point" during latency. This set point predicts the time of onset of clinical disease. With less than 1000 copies/ml of blood, disease will probably occur with a latency period of more than 10 years. With less than 200 copies/ml, disease does not appear to occur at all. Most patients with more than 100,000 copies per ml, lose their CD4+ cells more rapidly and progress to AIDS before 10 years. Most patients have between 10,000 and 100,000 copies per ml in the clinical latency phase.
d) Loss of CD4+ cells and abortion of the immune response. The major reason that the immune system fails to control HIV infection is that the CD4+ T helper cells are the target of the virus. Also follicular dendritic cells can be infected with HIV and these also diminish in number over time. Moreover, they present antigen to CD4+ cells and may bring the virus into contact with these cells at the time that they are stimulated to proliferate by antigen.
Note: During the course of infection, there is
a profound loss of the specific immune response to HIV:
i) because of the infection of the responding
CD4 cells. Thus there is clonal deletion leading to tolerance. The cells that proliferate
to respond to the virus are infected and killed by it;
ii) epitope variation (see below) can lead to
escape of HIV from the immune response;
iii) activated T cells are susceptible to
apoptosis. Spontaneous apoptosis of uninfected CD4+ and CD8+ T cells
often occurs in HIV-infected patients by an ill-understood mechanism. Also (via a process
that involves the viral protein nef - see below), there appears to be specific
apoptosis of HIV-specific CD8+ cells;
iv) the number of follicular
dendritic cells falls over time, resulting in diminished capacity to
stimulate CD4+ cells
There is thus a relentless decline of CD4+ cells with especially a loss of those specific to HIV. This occurs from the very beginning of infection and is permanent. Near the end stage of AIDS, CD8+ cells also decline precipitously.
e) Onset of AIDS. The period of clinical latency varies in length from as little as 1-2 years to more than 15 years but, eventually, the virus can no longer be controlled as the virus and cytotoxic T (CD8+) cells destroy helper CD4+ (T4) cells. Ironically, the killer cells needed to control HIV also damage the helper T cells that they need to function efficiently. With the lack of CD4+ cells, new cytotoxic T cell responses cannot occur as helper cells are lacking and such new responses are demanded as the virus mutates. As the T4 cells fall below 200 per cu mm, virus titers rise rapidly and immune activity drops to zero. It is the loss of immune competence that enables normally benign opportunistic parasites such as fungi or protozoa to cause infections. Once AIDS develops, patients rarely survive more than two years without chemotherapeutic intervention. (See anti-HIV chemotherapy section). There is considerable variability at this stage. Some patients with clinical AIDS do survive for several years while others who appear relatively healthy can suddenly succumb to a major opportunistic infection. It is the onset of HIV-associated neoplasm and opportunistic infections that defines AIDS proper. At this stage, also, syncytium-inducing HIV appear in many (about half) AIDS patients (see below). These are more CD4+ cell tropic than the initially infecting HIV and this switch contributes to the rapid loss of CD4+ cells in later stages of the disease.
Note: A phase of HIV infection, AIDS-related
complex (ARC), used to be defined. This is now little used. It is the phase
of disease that lacks the neoplasms and opportunistic infections that are the
definition of AIDS. Patients at this stage of the disease show weight loss and
fatigue together with fungal infections of the mouth, finger and toe nails.
 Despite possible co-factors associated with
lifestyle, HIV infected persons progress to AIDS at a remarkably similar rate.
The mean
time from seroconversion to onset of disease is approximately 9 years.
Perinatally-infected infants progress faster. Signs of AIDS can be seen by 5
months in more than 80% of seropositive children. About half die by nine years
of age. In neonates, the level of viral RNA rises rapidly in the first few
months of life but does not decline as rapidly as is seen in adults. The
decline that occurs, takes place over a period of a year or more. This presumably
reflects the less effective infant immune system. As in adults, the level of HIV
RNA predicts the rapidity of progression to AIDS.
Despite possible co-factors associated with
lifestyle, HIV infected persons progress to AIDS at a remarkably similar rate.
The mean
time from seroconversion to onset of disease is approximately 9 years.
Perinatally-infected infants progress faster. Signs of AIDS can be seen by 5
months in more than 80% of seropositive children. About half die by nine years
of age. In neonates, the level of viral RNA rises rapidly in the first few
months of life but does not decline as rapidly as is seen in adults. The
decline that occurs, takes place over a period of a year or more. This presumably
reflects the less effective infant immune system. As in adults, the level of HIV
RNA predicts the rapidity of progression to AIDS.
There are a variety of factors that determine progression of an HIV infection to clinical AIDS disease (Information Box)
Why Mosquitoes Cannot Transmit the AIDS Virus
HIV load but not CD4+ T cell number is a good indicator of rapid progression to AIDS
The onset of AIDS occurs on average 10 to 11 years after infection but in the absence of treatment some individuals progress much faster. About 20% of patients exhibit AIDS symptoms within 5 years of infection while others remain disease free much longer than average. The onset of AIDS correlates with the diminution of the number of CD4+ T cells but the major loss of T cells occurs late in infection. A marker that could predict the prognosis for AIDS progression early in the disease would be useful. Not surprisingly, the initial baseline viral load (that is when the patient is first monitored for virus number) is a good predictor of the time it will take for disease to appear. CD4 cell number is not a good predictor.
 Relationship between baseline viral load and progression to
AIDS. For quartiles from the lowest to the highest viral load at
time of entry into the study, the percentages of patients that progressed to
AIDS in 5 years were 8, 26, 49 and 62 (data from Mellors et al. Science 272:
1167-1170)
Relationship between baseline viral load and progression to
AIDS. For quartiles from the lowest to the highest viral load at
time of entry into the study, the percentages of patients that progressed to
AIDS in 5 years were 8, 26, 49 and 62 (data from Mellors et al. Science 272:
1167-1170) |
 Relationship between baseline viral load and median survival time. The
median time for the patients in each of these viral load categories was more
than 10 years, 7.7 years, 5.3 years and 3.5 years
Relationship between baseline viral load and median survival time. The
median time for the patients in each of these viral load categories was more
than 10 years, 7.7 years, 5.3 years and 3.5 years |
|
Co-factors in AIDS--The enigma of Kaposi's sarcoma
Kaposi's sarcoma (KS) is often a corollary of HIV infection in gay men (over 20% suffer from KS and HIV-positive patients are at 20,000 fold increased risk of KS compared to general population). Clinically, most KS is so indolent that many affected individuals die of other unrelated causes. It does, however, seem that the AIDS-associated form is more progressive involving many sites (skin, lymph nodes, lungs, intestine). This form of Kaposi's sarcoma, so typical of the progression of AIDS in gay men, is not found in some other populations that are HIV-positive. It has long been suspected, therefore, that Kaposi's is not the direct result of HIV but some co-factor that is activated in the presence of HIV. This now seems to be the case. In 1994, a Kaposi's sarcoma-associated herpes virus (KSHV or human herpes virus-8) was identified. In AIDS patients, antibodies against KSHV are common only in those that have KS or those that will eventually get KS (greater than 80% of this population). In contrast, blood samples from infected hemophiliacs show no anti-KSHV antibodies..
It is not known why the virus has established itself in primarily the gay population (if indeed it has) when most herpes viruses are widely disseminated throughout the population. It should be noted that there are some investigators who do not find KSHV to be so restricted in its range
AIDS is currently defined in persons older than 13 years as the presence of one of 25 conditions indicative of severe immunosuppression or HIV infection in an individual with a CD4+ cell count of <200 cells per cubic mm of blood. AIDS is therefore the end point of an infection that is continuous, progressive and pathogenic. With the prevalence of HIV in the developing world, HIV and its complications will be with us for generations.
Approximately 8500 new HIV infections occur daily around the world and over 90% of these are in developing countries. One thousand are in children less than 15 years of age. Of adult infections, 40% are in women and 15% in individuals of 15-25 years of age. Perinatal infection is now resulting in a large number of children being born with HIV. 30-50% of mother to child transmissions of HIV results from breast feeding and about a quarter of babies born to HIV-infected mothers are themselves infected.
From CDC: As of December 1997, 641,086 Americans had been reported with AIDS. At least 385,000 of them have died. Prior to the introduction of combination therapies for HIV, AIDS incidence was increasing at a rate of less than 5% each year. Partly as a result of prevention efforts targeting those at highest risk, the epidemic had slowed considerably from the early years when increases were 65% to 95% each year. In 1996, estimated AIDS incidence dropped for the first time, declining 6%. Deaths among people with AIDS also declined for the first time in 1996, dropping 25%.
In 1996, an estimated 242,000 people were living with AIDS in the United States, an increase of almost 12% since 1995. If declines in AIDS cases continue, there will also be an increase in HIV prevalence, pointing to an increased need for both prevention and treatment services. Estimates suggest that 650,000 to 900,000 Americans are now living with HIV, and at least 40,000 new infections occur each year.
Note: As more and more people survive with an HIV infection because of successful chemotherapeutic intervention, the number of infectious people in the population is rising even though fewer people are dying of AIDS.
AGE |
# of AIDS CASES |
Under 5: |
6,343 |
Ages 5 to 12: |
1,743 |
Ages 13 to 19: |
3,130 |
Ages 20 to 24: |
22,953 |
Ages 25 to 29: |
88,415 |
Ages 30 to 34: |
146,712 |
Ages 35 to 39: |
143,381 |
Ages 40 to 44: |
103,660 |
Ages 45 to 49: |
58,516 |
Ages 50 to 54: |
30,789 |
Ages 55 to 59: |
17,251 |
Ages 60 to 64: |
9,662 |
Ages 65 or older: |
8,530 |
In Africa (mostly sub Saharan), there are more than 21 million people with HIV infection and about 1 million new cases of AIDS per year. In 1996, South east Asia had 183,000 new cases and 5.2 million people with HIV. (For further global figures, go here)
AIDS is responsible for a decrease in life expectancy and increase in child mortality. Child mortality rates in East Africa will double by 2010 and adult life expectancy has already declined by 2 years in that region.
HIV infections have leveled off in the west and
the wave of infections threatening to affect western heterosexuals has not materialized.
However, this is not the case elsewhere and there have been huge increases in southern
Asia and southern Africa.
 HIV prevalence among pregnant women in South African provinces 1990-97. Several countries in sub-Saharan Africa report infection rates
of 20%, especially in urban areas. In some Kenyan and Zambian towns, 1 in 5 girls is HIV-positive by the age
of 20 (Information
Box). In men over
25, the percentage who are HIV-infected can be as high as 40%.
HIV prevalence among pregnant women in South African provinces 1990-97. Several countries in sub-Saharan Africa report infection rates
of 20%, especially in urban areas. In some Kenyan and Zambian towns, 1 in 5 girls is HIV-positive by the age
of 20 (Information
Box). In men over
25, the percentage who are HIV-infected can be as high as 40%.
In Botswana, the proportion of the adult population living with HIV has doubled
over the last five years, with 43% of pregnant women testing HIV-positive in
1997 in the major urban center of Francistown. In Zimbabwe, one in four adults
in 1997 were thought to be infected.
Current estimates are that more than
34 million people
will have HIV infection by 2000 (for the present number go here)
For the latest WHO statistics go here
For WHO report (June 1998) on HIV/AIDS statistics go here
(requires Acrobat)
It is likely that other factors than the presence of HIV influence the course of the disease but there is no strong evidence that there is any other specific infecting agent than HIV in AIDS (Please see appendix 2 for further details).
 Current HIV infection statistics by continent
Current HIV infection statistics by continent
AIDS cuts life expectancy in sub-Saharan Africa by a quarter
Two types of HIV can be distinguished genetically and antigenically. HIV-1 is the cause of the current pandemic while HIV-2 is found in west Africa but rarely elsewhere. The HIV-2 type is closely related to simian immunodeficiency virus (SIV) found in west Africa. There are also 10 different HIV-1 subtypes. The major one in the US and Europe is type B. In some countries, mosaics between different subtypes have been found (See Success Of Vaccines Against AIDS Cast In Doubt)
 HIV subtypes
by continent
HIV subtypes
by continent
Long-term non-progressors and exposed-uninfected individuals
Ever since the AIDS epidemic began there have been people who are clearly exposed to the virus who seem to exhibit no symptoms and normal CD4+ T cell counts. These are called long term non-progressors: People who have been infected with HIV for more than 7 years, who have stable CD4+ T cell counts above 600 per cu mm and have no history of symptoms and have not been taking anti-retroviral drugs. Their lymph node structure seems normal.
The CD4+ T lymphocytes of these patients fall after primary infection and seroconversion but remain at normal levels thereafter, in some cases up to 15 years. This seems to be a heterogeneous group of people whose long-term non-progressive disease results from a robust CD8+ T cell immune response against HIV, a poorly replicative virus, or mutations in co-receptors that HIV needs, along with CD4 antigen, to enter the cell.
As noted below (see section on co-receptors), a chemokine receptor on the surface of macrophages and activated T helper CD4+ cells was targeted by researchers as a co-receptor for HIV because of its known binding to three chemokines which seem to block infection. The nature of this co-receptor may be one explanation of the people who are exposed repeatedly to HIV but remain uninfected. It has been found that the cells of some of non-progressors are very resistant to HIV infection because they have mutant chemokine receptors. The most common of these is a 32 base pair deletion that prevents surface expression of a chemokine receptor known as CCR5.
CCR5 mutations are relatively rare. If 2 copies of the gene for CCR5 are defective, it is virtually impossible for virus to enter the cell and exposed patients are immune from HIV infection. Approximately 1 in 100 Caucasians have this double mutation. 17% have single defective gene. No African-Americans have so far been found with a double mutation but about 2% have single mutation. A single defective gene does not confer resistance but progress to disease is much less rapid. It is surprising that these people do not seem to have a reduced viral load or higher CD4 counts so the reason why these people progress better is unclear!
Recently another co-receptor, CCR2, has been identified. A heterozygous mutation in this may defer AIDS by an average of 2 to 4 years. This protective CCR2 mutation is present in all races in the U.S. at a frequency of about 20-25%. One quarter of long term survivors are CCR2 or CCR5 mutants
Other long term non-progressors appear to make elevated levels of the chemokines that bind to these receptors and perhaps this keeps the receptor blocked. This might lead to therapy in which the chemokine receptors of macrophages and T-cells are blockaded - as noted below, some chemokines are powerful suppressors of HIV infection in vitro.
Promoter Region Of CCR5 Gene May Control Progress Of HIV
Gene Marker Associated with Better Response to HAART
There are other suggestions concerning exposed-uninfected individuals. A study of Nairobi prostitutes, repeatedly exposed to HIV (25% or more of their clients are HIV positive), has shown that many of these women have been free of disease for more than 12 years and seem to be completely resistant to infection. There seem to be associations between their resistance to infection and their class I and class II MHC (HLA) haplotypes. The strongest association of protection is with HLA-A*6802, A*0202 and B18. These women have mounted a very strong CTL response that is likely to mediate protection. It is possible that these particular class I MHC antigens allow a very efficient CTL response. Alternatively, they may present epitopes that are highly conserved between different HIV-1 variant strains. For example, one epitope to which there is a strong CTL response in these women is that presented by B18. This epitope is found to be located in a highly conserved part of HIV p24 protein. It appears to be conserved because it is very important in the assembly of the virus. Another important epitope is presented by HLA-A*6802 and this is in the protease. The protease may not be able to bear much mutation in this region without losing enzymic activity and so the virus cannot escape the immune response by mutation.
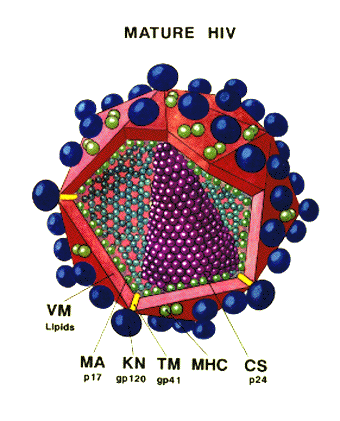
HIV is a retrovirus with a similar structure to other retroviruses.
 Diagram of the protein locations in HIV
(Click to enlarge)
Diagram of the protein locations in HIV
(Click to enlarge)
Surface structures
Viral membrane: This is host-derived as a result of budding from the cell surface. Some host proteins become incorporated into the viral membrane
Surface glycoprotein: Gp160 is encoded by the env gene. Gp160 is cleaved after translation by host enzymes in the Golgi Body to form Gp120 (SU) and Gp41 (TM). Gp 41 is embedded in the membrane, Gp120 is not but is held to Gp41 by non-covalent interactions. It is easily shed from the virus particle. Note: Gp120 and Gp41 are made from a single polypeptide. There is a large number of sugar chains on gp120 (Problem for vaccine). Gp120 is the protein that interacts with a receptor on the cell to be infected. Gp41 is the fusogen that is exposed after Gp120 has bound to the cell.
Internal structural proteins: These are all encoded in the gag gene. P17 lines the inner surface of viral membrane to which it is attached by covalently bound myristic acid. Other proteins associated with the nucleocapsid include p24 and p9. The group-specific antigen is made as a polyprotein and cleaved after budding of the virus by a virally-encoded protease encoded by the pol gene.
Other internal proteins: These are encoded by the pol gene. They are enzymes that participate in integration and replication: reverse transcriptase, integrase, protease.
Genome: Like other retroviruses, the genome is diploid positive sense RNA
Cells that are infected by HIV

CD4+ (T4) helper lymphocytes are the most studied but monocytes/macrophages are also infected and may provide an important reservoir. HIV also infects oligodendrocytes, astrocytes, neurones, glial cells. The virus destroys T4 cells specifically, causing profound immuno-suppression. Other cells tend to harbor the virus without lysis.
Follicular dendritic cells (FDCs) may also be involved. When virus enters the healthy lymph node, it is trapped by FDCs so that when T cells reach this region of the body (approx 20% of an individual's T cells go through this area daily), many are infected. Thus, FDCs serve as a reservoir for further infection.
CD4 antigen is the HIV receptor
![]() The apparent specificity of CD4+ cell
infection observed initially, together with the observation that T4 cells are those
that are depleted in disease (indeed, the course of disease in the patient is followed by
CD4+ cell levels), suggested that CD4 antigen might be the receptor for the
virus. This was demonstrated by transfecting CD4 antigen gene into CD4-
human cells (such as HeLa cells) and showing that they acquired the property of being able to be infected
by HIV.
The apparent specificity of CD4+ cell
infection observed initially, together with the observation that T4 cells are those
that are depleted in disease (indeed, the course of disease in the patient is followed by
CD4+ cell levels), suggested that CD4 antigen might be the receptor for the
virus. This was demonstrated by transfecting CD4 antigen gene into CD4-
human cells (such as HeLa cells) and showing that they acquired the property of being able to be infected
by HIV.
A co-receptor for infection by HIV
However, something more appears to be necessary as this experiment only works with human cells; for example, mouse cells transfected with CD4 antigen gene are not infected by HIV.
It was also discovered that some strains of HIV (those adapted for life in transformed T cells) could replicate in activated human T cells but not in monocytes or macrophages. Conversely, those adapted for life in macrophages could not replicate in transformed T cells. Yet both macrophages and T4 cells possess CD4 antigen. The differences in tropism of the viral strains mapped to the V3 region of Gp120 suggesting that molecules other than CD4 antigen have an important role in binding and this role is CD4+ cell type-specific.
Chemokine receptors seem to be the key to the gateway of the cell-- a family of proteins on the surface of immune cells.
CCR5. Several laboratories identified an
essential co-receptor for those HIV strains involved in critical early stages of
infection (these are the macrophage-tropic strains). All of these studies found CCR5
as the partner of CD4 in allowing entry into macrophages.
 The discovery of this molecule
as the co-receptor came from previous studies that showed that three chemokines secreted
by CD8+ T lymphocytes (called RANTES, MIP-1a and MIP-1b), which are involved in
inflammatory responses, are powerful suppressors of HIV infection, and especially
macrophage-tropic HIV. Of course, this suggested that the suppression of infection might
be because the chemokines bound to the cell surface and blocked a receptor that HIV needed
for entry. Suspicion fell on CCR5 because it was known to be the receptor that binds all
three of the above chemokines.
The discovery of this molecule
as the co-receptor came from previous studies that showed that three chemokines secreted
by CD8+ T lymphocytes (called RANTES, MIP-1a and MIP-1b), which are involved in
inflammatory responses, are powerful suppressors of HIV infection, and especially
macrophage-tropic HIV. Of course, this suggested that the suppression of infection might
be because the chemokines bound to the cell surface and blocked a receptor that HIV needed
for entry. Suspicion fell on CCR5 because it was known to be the receptor that binds all
three of the above chemokines.
CXCR4 also known as fusin (a G protein-coupled receptor whose ligand is a B cell stimulatory factor--it is called fusin because it promotes the infection/fusion of CD4+ cells) is also a co-receptor for HIV in otherwise non-infectable CD4+ cells. The amount of fusin on the cell surface may explain differences in tropism and fusin seems to be more active in T cells than in macrophages. Note the fusin gene most closely resembles the gene for IL-8 receptor (also a chemokine receptor)
Recently another co-receptor (CCR2) has been found.
Complexes of pieces of CD4 and Gp120 also bind to CCR5 on CD4- cells. This explains why soluble CD4 actually enhances HIV infectivity and does not block it. It seems that gp120 binds CD4 and undergoes a conformational change that increases its affinity for the chemokine receptor. The binding of the chemokine receptor causes a conformational change in the gp41 fusion protein of HIV that allows fusion of the viral membrane with the membrane of the cell to be infected. In fact, it is really the chemokine receptor that is the primary receptor for HIV and the role of CD4 is to concentrate virus at the cell surface and facilitate interaction with the chemokine receptor. In contrast to examples of CD4-independent HIV entry into cells, there are (so far) no examples of entry independent of chemokine receptors.
These co-receptors may explain the phenotypic switch during infection (see below). Changes in the amino acid sequence of gp120 occur in the progression of the disease. It is likely that HIV uses CCR5 in the early stages of disease and then switches to CXCR4 perhaps avoiding the suppressive activity of chemokines. This also explains the transition from non-syncytium-inducing to syncytium inducing phenotype. Note: CXCR4 and CCR5 are members of a very large family (thousands) of receptors and the spread of HIV through subtypes of T cells may reflect subtle changes on the variable loops of gp120 allowing the infection of new CD4+ cells with different receptors. This may also be one reason why so few CD4+ cells appear to be infected at any one time.
PowerPoint slides of the entry of HIV (go here)
Shockwave movie of entry of HIV (go here) Requires Shockwave plug-in
Some CD4-negative cells can be infected by HIV
It was originally thought that only cells that have CD4 antigen are infected. Although CD4 protein had not been demonstrated on some infectable cells, it was thought to be present in low amounts and the mRNA could be detected in most infectable cells. Specificity to CD4 positive cells reflects the specific binding of gp120 to CD4. It is now known, however, that some non-CD4 cells, such as those in brain and intestine, can be infected in a via a galactocerebroside receptor. Other cells can be infected in a different way; for example, in macrophages (see below), an Fc or complement receptor may be used. In these cases, the HIV must be bound by anti-HIV antibodies that interact with receptors on the cell surface. Thus anything that can up-regulate Fc receptors on macrophages will augment infection.
Entry into cell: pH-independent fusion with plasma membrane.
No pH-dependent conformational change in a viral membrane protein is necessary for fusion between the viral membrane and the membrane of the cell to be infected. Thus, no entry into lysosomes is required.
Remember from the lecture on herpes virus: This sort of fusion of virus with the plasma membrane is associated with fusions of infected cells to form syncytia. Syncytium formation is also a characteristic of HIV infection. This has profound significance for spread of infection between cells without any free virus. This means that virus may spread from cell to cell so that immune system circulatory antibodies cannot have any effect (problem for vaccine). Not only will a vaccine have to be able to destroy the virus, it will also have to be able to destroy infected cells. Gp41 is the fusogen. Syncytia are most often seen in brain.
 Multinucleated cell (syncytium) in touch preparation from cut surface of enlarged lymph node from patient with HIV-1 infection. Cell fusion producing a large multinucleated cell is a viral cytopathic effect characteristic, but not diagnostic, of infection by HIV-1. Giemsa stain.
Lymphadenopathy smear. CDC/Dr. Edwin P. Ewing, Jr. epe1@cdc.gov
Multinucleated cell (syncytium) in touch preparation from cut surface of enlarged lymph node from patient with HIV-1 infection. Cell fusion producing a large multinucleated cell is a viral cytopathic effect characteristic, but not diagnostic, of infection by HIV-1. Giemsa stain.
Lymphadenopathy smear. CDC/Dr. Edwin P. Ewing, Jr. epe1@cdc.gov
Reverse transcription and integration This
is similar to other retroviruses. HIV uses reverse transcriptase imported during infection
as part of the virus.
Formation of polyproteins and their cleavage: Click on thumbnail to enlarge
 Assembly of new
virus takes place at the membrane of the host cell. Three types of protein make up the virion. These are the
membrane protein complex (gp120 and gp41 - originally derived from gp160) plus two internal
precursor proteins, the GAG polyprotein and the GAG/POL polyprotein (the latter
is the result of a frame shift that allows the ribosome to continue translation
from the GAG gene into the POL gene)
Assembly of new
virus takes place at the membrane of the host cell. Three types of protein make up the virion. These are the
membrane protein complex (gp120 and gp41 - originally derived from gp160) plus two internal
precursor proteins, the GAG polyprotein and the GAG/POL polyprotein (the latter
is the result of a frame shift that allows the ribosome to continue translation
from the GAG gene into the POL gene)
 The proteins
aggregate at the cell membrane and the membrane pinches off. The larger internal precursor
draws two strands of the positive strand RNA into the nascent virion and the protease
(part of the larger internal precursor protein) cuts itself free
The proteins
aggregate at the cell membrane and the membrane pinches off. The larger internal precursor
draws two strands of the positive strand RNA into the nascent virion and the protease
(part of the larger internal precursor protein) cuts itself free
 The protease completes the cleavage of the larger
internal precursor to liberate other enzymes (reverse transcriptase, integrase and more
protease).. The protease cleaves the remainder of the larger internal precursor and the
smaller internal precursor into structural proteins. p24, p7 and p9 form the bullet-shaped
core
The protease completes the cleavage of the larger
internal precursor to liberate other enzymes (reverse transcriptase, integrase and more
protease).. The protease cleaves the remainder of the larger internal precursor and the
smaller internal precursor into structural proteins. p24, p7 and p9 form the bullet-shaped
core
Note: The GAG and GAG/POL fusion proteins are made in ratio of about 20:1. After the virus has budded from the cell, the protease cuts itself free and cuts up rest of proteins in GAG or GAG/POL, releasing the various structural proteins and reverse transcriptase. This specific protease is vital as proteins are not functional unless separated. This specific HIV protease has become a good candidate for a site for an anti-HIV drug (see below and chemotherapy lecture notes). GAG/POL and GAG are attached to membrane via a fatty acid that is covalently bound. The cleavages result in p17 remaining attached to the membrane. Note gp160 is cut to gp120 and gp41 by a host enzyme in the Golgi apparatus since it gets to the cell membrane via the exocytic pathway.
 Transmission electron micrograph of HIV-1, budding and free
(from Centers for Disease Control)
Transmission electron micrograph of HIV-1, budding and free
(from Centers for Disease Control)
LATENCY OF HIV
Most infected CD4+ cells are rapidly killed by the virus but these are initially replaced. CD4+ lymphocytes replicate HIV only when they are activated by contact with an antigen when they produce prodigious amounts of virus that lead to cell lysis and cell death. In contrast to most viruses, cell division is not essential to HIV replication. [Two viral proteins (Vpr and one of the GAG proteins) have nuclear localization signals so that mitosis (which removes the nuclear membrane barrier to penetration of viral DNA into the chromosomes) is not needed.] However, a resting T cell can take its parasite only so far, that is to the pre-integration complex. Thus a variety of viral and bacterial infections (and unfortunately vaccinations) can markedly increase HIV load in plasma.
In contrast, macrophages do not show latency. They bud virus all the time.
Note: two forms of latency that are often confused. Cellular latency refers to the fact that in activated CD4+ T helper cells, the virus can become dormant when a small proportion of these cells revert back to the resting memory state until the cell is activated again. This may be a few hours or days or very much longer in a very small minority of cells. Clinical latency refers to fact that symptoms of HIV infection do not manifest themselves as AIDS for many years.
What is the explanation of the complex life history of HIV? How can it remain dormant and then break out of the cell in prodigious quantity?
The amount of HIV in the body is high just after infection but the immune system deals effectively with it. The virus declines and there is effective control for years. CD4+ cells are lost during this period as some virus is around to infect them, leading to more shed virus. An equilibrium is set up between cell loss and the ability of the body to replace them. Initially, cell replacement is very effective, then the balance shifts and the virus gains the upper hand. HIV begins to replicate wildly in the cells and the remaining CD4 cells disappear.
Since HIV has a more complex life cycle that simple retroviruses such as RSV and it appears that HIV can control its replication in a more complex fashion, we might expect more genetic information but this is not so.
THE GENOME OF HIV

HIV genome is 9749 nucleotides-- about the same size as any other retrovirus, for example Rous sarcoma virus (RSV).
But the genome of HIV is more complex than RSV since it has extra open reading frames that clearly code for small proteins. Some of these are protein synthesis-controlling proteins.
The HIV genome has nine open reading frames but 15 proteins are made in all
The GAG/POL polyprotein and the ENV polyprotein are cleaved into several
proteins:
GAG is cleaved to: MA (matrix), CA (capsid), NC (nucleocapsid), p6
POL is cleaved to: PR (protease), RT (reverse transcriptase), IN (integrase)
ENV is cleaved to: SU (gp120) and TM (gp41)
Of the other proteins, three are incorporated into the virus (Vif, Vpr and Nef) while three are not found in the mature virus: Tat and rev are regulatory proteins and Vpu indirectly assists in assembly
TAT: Trans-Activator of Transcription
REV: Regulator of Virion protein expression
NEF: Negative Regulatory Factor
VIF: Virion Infectivity Factor
VPU: Viral Protein U
VPR: Viral Protein R
These genes encode small proteins. They overlap with the structural genes (especially ENV) but are in different reading frames. Note that some are encoded in two exons (unlike the structural genes) and therefore their mRNAs can be derived by alternative splicing of structural gene mRNAs. This is rather important to the way in which the levels of these are controlled. Mutants in the TAT and REV genes show that they are both vital to any production of virus.
TAT gene product binds to a sequence in all of the genes and positively stimulates transcription. It is thus a positive regulator of protein synthesis, including its own synthesis.
REV binds to an element only in the mRNA for structural proteins (GAG/POL/ENV) and regulates the ratio of GAG/POL/ENV to non-structural, controlling protein (TAT/REV) synthesis. When REV levels are high, structural protein synthesis rises and controlling protein synthesis falls. Thus REV inhibits its own production and that of TAT.
The normal result is homeostasis, low or non-existent virus production and latency in the resting CD4 cell.
Note: HIV's lifestyle leads to an inherent problem. It uses genomic RNA as a messenger RNA. This RNA is unspliced and the nucleus has a mechanism to prevent unspliced mRNAs from leaving the nucleus and being translated.. It is the function of rev to overcome this problem.
Despite its small size NEF has several functions.
a) Homeostasis leads to several problems for the parasitic provirus: super-infection by other HIV particles--too many parasites may kill the cell. Probably more importantly, virus bound via CD4 antigen at the cell surface or free gp120 bound to CD4 antigen at the cell surface may result in the cell being subject to an immune attack. In addition, loss of CD4 will enhance the escape of virus produced by the cell since they will not remain bound to the cell surface (see also Vpu, below).

 The translation of the NEF gene as a
result of the first infecting virus (NEF gene is translated early in infection) causes the internalization of CD4 antigen from the
cell surface and its destruction in lysosomes. Thus no more HIV or gp120 can bind!
The translation of the NEF gene as a
result of the first infecting virus (NEF gene is translated early in infection) causes the internalization of CD4 antigen from the
cell surface and its destruction in lysosomes. Thus no more HIV or gp120 can bind!
b) By a different mechanism from its down regulation of CD4 antigen, NEF reduces surface expression of MHC class I molecules. This alters antigen presentation by the infected cell and is proposed to protect the infected cell from attack by cytotoxic T cells
c) The name, NEF, comes from negative factor. Originally, it seemed that virions that lacked NEF grew better than wild type. Now the consensus is for the opposite, that is that virus produced in the presence of NEF is a little more infectious than virus produced in its absence.
d) It is found that NEF is important for HIV replication in vivo but there seems to be little effect of NEF in an in vitro cell culture situation. Why the is so has long been obscure. Recently, this problem seems to have been solved and the answer comes from the macrophages which are changed in two ways when they are infected with a NEF-expressing HIV (remember that macrophages are the cells that bring HIV into the body and the initial strains of HIV in an infected patient are macrophage-tropic).
 The infected macrophages secrete MIP-1alpha and MIP-1beta which are two
chemokines that incidentally bind to the co-receptors for HIV infection of
macrophages. These chemokines have another function.
The infected macrophages secrete MIP-1alpha and MIP-1beta which are two
chemokines that incidentally bind to the co-receptors for HIV infection of
macrophages. These chemokines have another function.
 They cause
resting CD4+ T cells to migrate (undergo chemotaxis) towards the
macrophages. This is important in the in vivo situation since, initially,
HIV infected cells are not very numerous and uninfected T cells may not be in
the vicinity of the infected cells. Moreover, HIV does not have a very long half
life in the circulation before becoming non-infectious. Migration increases the
probability that the T cells will encounter infected macrophages before the
virions leave the reticuloendothelial system. This explains why NEF seems
not to be of much consequence in cell culture where the cells are already close
together. The infected macrophages do something else. They make a factor that
has not yet been identified that activates the resting T cells that they have
attracted allowing them to be productively infected and to shed new virus
(Remember, lentiviruses, unlike most other retroviruses can infect non-dividing
cells. Normally, retroviruses can only enter the nucleus and integrate during
mitosis when the nuclear membrane is broken down; however, proteins of
lentviruses such as HIV have nuclear targeting signals that allow the nucleocapsid to find
the nuclear pore. Thus, the virus can integrate into the host cell chromosome
where it can now code for more viral RNA and protein. HIV, unlike some other
lentiviruses, does not transcribe its genome to RNA in resting T cells since the
activation of the promotor in the LTR requires transcription factors that are
only made when a resting T cell becomes activated). These finding explain
why macrophages are vital for the spread of HIV.
They cause
resting CD4+ T cells to migrate (undergo chemotaxis) towards the
macrophages. This is important in the in vivo situation since, initially,
HIV infected cells are not very numerous and uninfected T cells may not be in
the vicinity of the infected cells. Moreover, HIV does not have a very long half
life in the circulation before becoming non-infectious. Migration increases the
probability that the T cells will encounter infected macrophages before the
virions leave the reticuloendothelial system. This explains why NEF seems
not to be of much consequence in cell culture where the cells are already close
together. The infected macrophages do something else. They make a factor that
has not yet been identified that activates the resting T cells that they have
attracted allowing them to be productively infected and to shed new virus
(Remember, lentiviruses, unlike most other retroviruses can infect non-dividing
cells. Normally, retroviruses can only enter the nucleus and integrate during
mitosis when the nuclear membrane is broken down; however, proteins of
lentviruses such as HIV have nuclear targeting signals that allow the nucleocapsid to find
the nuclear pore. Thus, the virus can integrate into the host cell chromosome
where it can now code for more viral RNA and protein. HIV, unlike some other
lentiviruses, does not transcribe its genome to RNA in resting T cells since the
activation of the promotor in the LTR requires transcription factors that are
only made when a resting T cell becomes activated). These finding explain
why macrophages are vital for the spread of HIV.
Note: NEF seems to be able to cause the activation of resting T cells but, in
vivo, HIV cannot productively replicate in resting T cells so NEF cannot be
made. The above observations solve this conundrum. Macrophages are infected by
HIV and make NEF without any activation process. As a result they make
factors that activate resting T cells that can now support a productive
infection!
After activation the virus faces another problem: CD4 antigen and gp120 are being made in the endoplasmic reticulum of the same cell. They are likely to bind to one another before reaching the plasma membrane and such complexes are usually targeted by the cell for degradation. To stop this unfortunate state of affairs, another of the small HIV proteins (VPU) promotes the proteolysis of the CD4 antigen of the host cell as it is made!
See also p6 (Information Box), Vif (Information Box), Vpr (Information Box)
One HIV Gene Kills All Tumors In Lab Tests
(Vpr)
Latency is broken when the virus proliferates and this seems to occur when the CD4 cell is stimulated during an antigenic response. How? One factor known to be involved is nuclear factor NF kappa B (NFkB, for short).
When a lymphocyte is stimulated by an antigen or by a cytokine binding to its surface receptor, it responds by proliferating. The signal to grow is relayed from the cell surface to the genes by transcription factors of which NFkB is one. The NFkB binds to a site in the promotor region of the genes that are specifically activated. The VERY SAME sequences are found in the HIV LTR!
Why is there a progressive, inevitable loss of the CD4+ helper T-cells? Why do CD8+ killer T cells disappear late in disease?
Why, when only 1 in 10,000 (early) or 1 in 40 (later) cells show productive infection, do all of the T4 cells disappear? It is still unclear why the CD4+ cells all disappear
Possibilities:
 a) Punctured membranes as virus buds (but the cell needs
to be infected)
a) Punctured membranes as virus buds (but the cell needs
to be infected)
 b) Syncytia formation (spread of virus to uninfected
cells)
b) Syncytia formation (spread of virus to uninfected
cells)
 c) Infected cell looks like the virus since it is
gp120 positive (and is therefore destroyed by cytotoxic T cells)
c) Infected cell looks like the virus since it is
gp120 positive (and is therefore destroyed by cytotoxic T cells)
d) Shed gp120/gp41 binds to uninfected cells via CD4 antigen. As a result, they are seen as infected and destroyed (see c)
e) AIDS-related cytotoxic antibodies may react with a specific antigen on surface of activated T4 cells.
f) AIDS might have an auto-immune component. In a normal antigenic response of T4 cells, CD4 interacts with type II histocompatibility antigen (MHC). Since gp120 also binds to CD4, the gp120 can mimic MHC and there appear to be regions of similar sequence. Thus anti-gp120 antibodies may turn out to be anti-MHC antibodies as well. (Might spell trouble for vaccine production).
g) Involvement of unknown T4 cell stem cell population? Or perhaps there is a subset of T4 cells that is vital to propagation of entire population of T4 cells
h) HIV proteins may alter T4 cell function. There is some evidence for this.
i) HIV protein (possibly NEF) may be super antigen.
j) HIV initiates apoptosis that in all T4 cells (this is normal in some T4 cells to overcome autoimmunity).
Some of the above may explain why only a minority of T4 cells appear to be infected at a given time yet all disappear in the later stages of the disease. It could also be that the virus switches from one T4 cell population to another as it switches its co-receptor (see above).
CD8+ cells are not infected by HIV (they do not have the CD4 receptor) and their numbers remain high during the course of the disease for many years. And then, until recently inexplicably, they rapidly die off. Until this point, the virus is controlled but now the virus seems to take over and reproduces in the remaining CD4 cells which also die. AIDS ensues. It appears that some of the HIV subtypes that occur late in infection prompt a mass apoptosis of CD8 cells. Although CD8 cells are CD4-, they do have CXCR4 co-receptor and HIV can bind to this (only the later syncytium-inducing strains do this). Since no CD4 antigen is present there is no infection but binding to CXCR4 does send a signal to the cell, the signal for apoptosis and mass CD8+ cell suicide ensues. It is interesting that the CD8 cells only die when macrophages are present.
How does this happen? It is now known that binding of strains of HIV that arise later in infection to the CXCR4 receptor sets in motion the tumor necrosis-alpha death transducing pathway.
 In macrophages, binding of a ligand to CXCR4 receptor on the cell surface
induces the expression of TNF-alpha. In CD8+ T cells, the same binding triggers
the expression of TNF-alpha receptor II.
In macrophages, binding of a ligand to CXCR4 receptor on the cell surface
induces the expression of TNF-alpha. In CD8+ T cells, the same binding triggers
the expression of TNF-alpha receptor II.
 When such a macrophage and CD8+ T cell come in contact, the TNF-alpha on the
macrophage binds to the receptor on the CD8+ T cell. This triggers an
apoptosis signal in the CD8+ T cell resulting in the vesiculation of the CD8+ T
cell.
When such a macrophage and CD8+ T cell come in contact, the TNF-alpha on the
macrophage binds to the receptor on the CD8+ T cell. This triggers an
apoptosis signal in the CD8+ T cell resulting in the vesiculation of the CD8+ T
cell.
 Macrophages then phagocytose the remains of the T cell. This explains why
macrophages have to be present for the CD8+ cells to die. Why would this happen
naturally? Why do chemokines act as death signals for CD8+ T cells? These cells
are killer cells and may cause serious trouble if they end up in the wrong
place. It is thought that chemokines direct CD8+ T cells to the fate of
macrophage-mediated death unless they reach their appropriate location.
Macrophages then phagocytose the remains of the T cell. This explains why
macrophages have to be present for the CD8+ cells to die. Why would this happen
naturally? Why do chemokines act as death signals for CD8+ T cells? These cells
are killer cells and may cause serious trouble if they end up in the wrong
place. It is thought that chemokines direct CD8+ T cells to the fate of
macrophage-mediated death unless they reach their appropriate location.
How HIV Virus Kills
Cells It Doesn't Even Infect
Other cells that are infected by HIV
Although CD4+ T4 cells are usually considered to be the most important cells in the course of AIDS and it is their loss that leads to immune suppression, other cells get infected. Macrophages are very important as they form a reservoir outside the blood and carry the virus into tissues. Non-proliferating mature macrophages can support HIV production for a long time without being killed. There is no latency in these cells, the virus just buds. Cytokine production by the infected macrophages is also aberrant leading to a variety of secondary effects. The slim disease that is characteristic of HIV infections in Africa may result from macrophage cytokine disruption. This wasting is very reminiscent of Visna in sheep and Visna infections involve the macrophages. Macrophages and macrophage-like cells are infected via CD4 antigen. Also since the virus raises good antibodies in the host, cells that express Fc or complement receptors will take up the virus.
Some CD4-negative cells become infected (epithelial cells of vagina and rectum), endothelial cells of brain capillaries, other cells of CNS. These may take up HIV via a galactocerebroside receptor. Dendritic cells may also be very important. These cells appear to trap virions and when the virus disappears from blood stream the dendritic cells of the lymph nodes may be the major site of the virus.
 Histopathology showing microglial nodule in brain of patient who died of AIDS. Brain microglial nodules are nonspecific microscopic brain lesions often associated with fatal HIV infection.
(CDC/Dr. Edwin P. Ewing, Jr. epe1@cdc.gov)
Histopathology showing microglial nodule in brain of patient who died of AIDS. Brain microglial nodules are nonspecific microscopic brain lesions often associated with fatal HIV infection.
(CDC/Dr. Edwin P. Ewing, Jr. epe1@cdc.gov)
Population polymorphism and HIV variants
Population polymorphism results from the high error rates of reverse transcriptase and RNA polmerase II which are used to replicate the viral genome. The error rate is 1 in 2000-10,000 nucleotides. This together with the high rate of CD4+ cell production and infection means that every possible single point mutation in the viral genome arises daily and almost 1% of all possible double mutations occur each day. As a result, the virus isolated from an AIDS patient is very different from the original infecting virus. Distinct sub-strains differ in cell tropism. Some form syncytia, some do not. The non-syncytium-inducing macrophage-tropic type is probably the infectious form (note: most vaccines made against syncytium-forming form). The major variable protein is gp120 and, within a single patient, HIV-1 commonly varies by 1-6% in the ENV gene. Although there are some conserved sites, mutations here presumably are non-viable (e.g. CD4 binding site). Also glycosylation masks these conserved sites (Masking and polymorphism are a problem for vaccine). Gp41 is not so glycosylated and fusion site needs to be conserved (this may be a possible vaccine site?).
Compared to variation within an individual, there is a lot greater variability around the world. HIV-1 genetic subtypes differ by up to 30% in amino acid sequence of ENV gene. There are at least 10 subtypes of HIV-1.
Not only is the reverse transcriptase mutation rate a problem. It is possible to be infected by different HIV-1 subtypes and cells become co-infected. Resultant viruses have one RNA from one subtype and one from the other. On later rounds of infection recombination occurs. It has been found that a recombinant subtype (HIV-1E) is spreading globally.
The impact of these evolving subtypes is great since they may affect the efficacy of tests for infected blood. Moreover, they have to be taken into account when thinking of a vaccine. There is also the possibility that there may be significant differences in the transmissibility of different subtypes (HIV-1 is much more transmissible than HIV-2)
Chemotherapy: Most anti-HIV drugs are toxic. In addition, anti-HIV chemotherapy will not stop infection and is unlikely to cure the infected host (though see chemotherapy lecture notes). The most we can hope for is suppression of virion production making AIDS a more tractable disease. Recently great strides towards this goal have made (Please see appendix 3).
Education: HIV is (fortunately) not highly infectious. It can be avoided by taking the correct precautions.
Vaccine: The best way to protect against an original infection. But HIV is a retrovirus and this poses enormous problems for vaccine development.
APPENDICES
Appendix 1: TOWARDS AN ANTI-HIV VACCINE
Appendix 2: DOES HIV CAUSE AIDS?
Appendix 3: THE CURRENT STATUS OF ANTI-HIV CHEMOTHERAPY (See, also, the chemotherapy lectures)
![]() Go here for the current status of specific anti-HIV drugs and links to drug data
sheets
Go here for the current status of specific anti-HIV drugs and links to drug data
sheets
![]() Return to the Microbiology and Immunology Department Site Guide
Return to the Microbiology and Immunology Department Site Guide
This page copyright
2000, The
Board of Trustees of the University of South Carolina
This page last changed on Wednesday, May 28, 2003
Page maintained by Richard Hunt
URL: http://www.med.sc.edu:85/hiv2000.htm
Please report any problems to rhunt@med.sc.edu