| MICROBIOLOGY AND IMMUNOLOGY ON-LINE |
Anti-coronavirus drugs
Drugs that inhibit RNA-dependent RNA polymerase
Most drugs that inhibit RNA virus replication have been developed as treatments for HIV, a retrovirus. This virus replicates its genome using reverse transcriptase, an RNA-dependent DNA polymerase. AZT (figure) was the first reverse transcriptase inhibitor that was successfully used in the treatment of HIV-AIDS and is a deoxynucleotide analog that is incorporated into viral DNA by reverse transcriptase and acts a chain terminator. AZT is a deoxyadenosine analog in which the sugar moiety is deoxyribose
Many zoonotic RNA viruses that infect humans do not have the nuclear phase of retroviruses and replicate entirely in the cytoplasm using a virus-encoded RNA-dependent RNA polymerase. Thus reverse transcriptase inhibitors would be unlikely to work and, as a result of the COVID-19 pandemic drugs, that inhibit RNA-dependent RNA polymerases are in development.
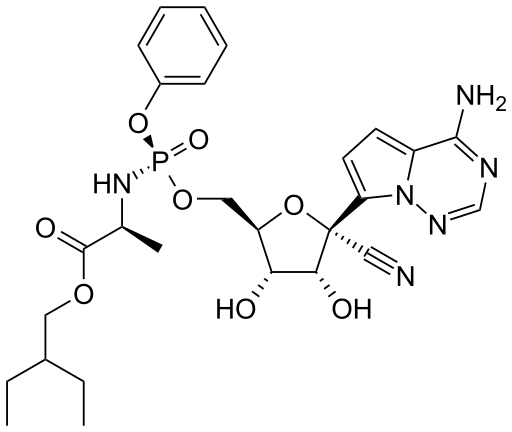
Remdesivir (GS5734)
An early drug to be tested against SARS-CoV-2 was remdesivir. It was originally developed as a treatment for Hepatitis C (a flavivirus), Ebola virus and Marburg virus (both filoviruses) but has been found to be highly active in cell culture studies against SARS-CoV-1, MERS CoV and SARS-CoV-2 (the causative agent of COVID-19). Thus, remdesivir is sometimes described as a pan-viral drug which is to be expected since all RNA viruses must encode their own RNA polymerase.
Remdesivir is an adenosine analog in which the sugar moiety is ribose. Like AZT, it is a prodrug that has to be metabolized into an active form (i.e. phosphorylated) before it can act as a chain-terminating substrate for the RNA polymerase.
As noted elsewhere Coronaviruses are the largest of the known RNA viruses and SARS-CoV-2 encodes a protein, NSP14 (ExoN), that acts as a proof reading exonuclease. Such an activity could be a problem using nucleoside chain terminator drugs. However, although it has been shown that a mutant mouse hepatitis virus that lacks ExoN is more sensitive to remdesivir, the drug inhibits the polymerase (NSP12) even in the presence of the proofreading activity and any resistance can be overcome by increased drug concentration. However, an initial Chinese trial in which 158 patients were randomly given remdesivir, and 79 received standard care with a placebo instead. There was no difference in recovery time between the groups. Just under 14% of those given remdesivir died, compared with 13% of those treated with the placebo. The trial was stopped early when 11.6% of patients on remdesivir experienced adverse effects compared to 5.1% on placebo.
Favipiravir (T-705, Avigan)
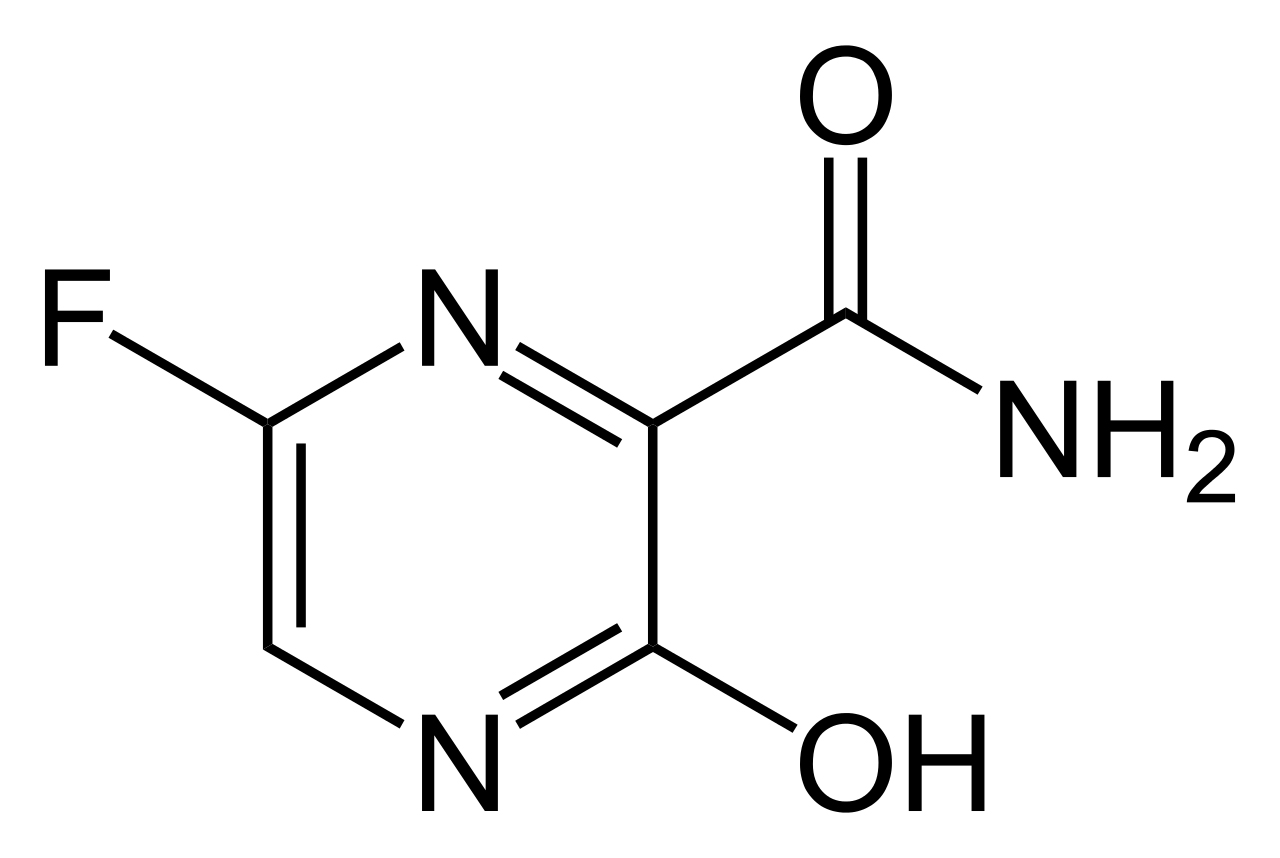
Favipiravir is a pyrazinecarboxamide derivative (6-Fluoro-3-hydroxy-2-pyrazinecarboxamide) and another inhibitor of the RNA-dependent RNA polymerase. It is a purine analog that was originally developed to treat influenza but was found to have low efficacy. It is a prodrug and a much simpler molecule that remdesivir. It is metabolized to its active form, favipiravir-ribofuranosyl-5'-triphosphate, by hypoxanthine guanine phosphoribosyltransferase which converts it into its ribose-5'-monophosphate form which is then converted to the triphosphate. It appears to compete with GTP in RNA synthesis and gives rise to lethal mutations in the viral RNA while having no effect on endogenous nuclear RNA synthesis.
Lysosomotropic drugs
Chloroquine and Hydroxychloroquine
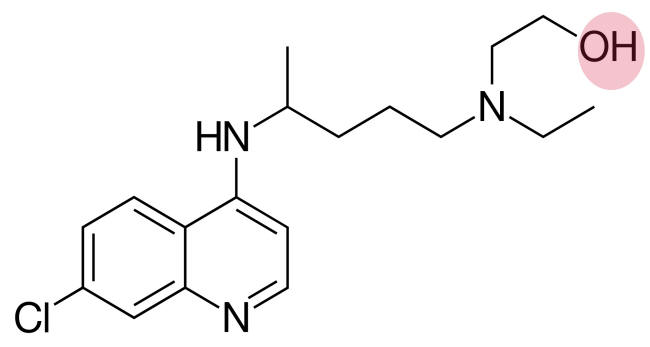
Chloroquine and hydroxychloroquine are widely used as anti-malarial drugs and also for the treatment of rheumatoid arthritis, lupus, and porphyria cutanea tarda. In the spring of 2020, a now retracted paper suggested the use of hydroxychloroquine in combination with the macrolide antibiotic azithromycin as a treatment for patients infected with SARS-CoV-2. Experiments in cultured Vero cells showed that hydroxychloroquine was more potent than chloroquine at inhibiting SARS-CoV-2 in vitro with a high selectivity index; as a result, numerous clinical trials of these old drugs alone or in combination with other drugs are being conducted. However, previous attempts to use them as anti-virals in vivo have shown mixed results and sometimes they have exacerbated the disease. Chloroquine inhibited Ebola virus replication in vitro but caused rapid worsening of Ebola infection in guinea pigs and made no difference to mortality in mice and hamsters. Similarly, these drugs worsened symptoms in chikungunya virus infections in monkeys. In April 2020, the first results of a trial of hydroxychloroquine showed that in a group of US veterans treated for COVOD-19, the risk of death from any cause was significantly greater in those receiving the drug compared to those who did not. In addition, a randomized trial in China showed no evidence of the efficacy of hydroxychloroquine.
Thus, what seem to be promising results in vitro (i.e. cell culture) are often not reflected in vivo. It should also be noted that while these drugs have been used clinically for a long time, especially as anti-malarials, they are not without serious side effects including inducing cardiac arrhythmias in genetically predisposed patients in whom the drugs block ion channels that are involved in heart beat control. Azithromycin was probably used in the original study to control bacterial infections and it can increase cardiovascular death when used in association with hydroxychloroquine in rheumatoid arthritis patients.
Although the chloroquines (4-aminoquinolines) have long been used as anti-malarials, their mechanism of action is still poorly understood. They are lysosomotropic agents that inhibit the acidification of late endosomes and lysosomes and may block entry of some viruses into the cytoplasm. This has been suggested for their effects on HIV replication. In the case of some coronaviruses and other viruses, an acidic environment is required for a conformational change in the virus surface protein (the S protein in the case of coronaviruses) so that the fusogen region of this molecule can promote the fusion of the viral envelope membrane with the endosomal/lysosomal membrane resulting in entry of the nucleocapsid into the cytoplasm. Another possibility suggested by molecular modeling, is that both drugs can bind sialic acid and gangliosides with high affinity. A ganglioside binding site was also identified on the SARS-CoV-2 S protein which may be involved in the attachment of the virus to lipid rafts in the cell plasma membrane as a preliminary to the attachment of the S protein to the ACE-2 receptor. Thus, it may be that the drugs, by binding gangliosides, inhibit the binding of the virus to cell surface receptors.