| MICROBIOLOGY AND IMMUNOLOGY ON-LINE |
DROGAS ANTI-CORONAVÍRUS
Drogas que inibem a RNA polimerase RNA-dependente
A maioria das drogas que inibem a replicação de vírus de RNA têm sido desenvolvidas como tratamentos contra o HIV, um retrovírus. Este vírus replica seu genoma usando a transcritase reversa, uma DNA polimerase dependente de RNA. AZT (figura) foi o primeiro inibidor de transcritase reversa que foi usado com sucesso no tratamento da HIV-AIDS e é um análogo de desoxiribonucleotídeo que é incorporado no DNA viral pela transcritase reversa e age como um terminalizador de cadeia. AZT e um análogo de desoxiadenosina no qual o açúcar é desoxiribose.
Muitos vírus zoonóticos de RNA que infectam humanos não têm a fase nuclear dos retrovírus e replicam inteiramente no citoplasma usando uma RNA polimerase dependente de RNA. Dessa forma, inibidores de transcritase reversa não seriam capazes de funcionar e, como resultado das drogas contra a pandemia do COVID-19 drogas que inibem RNA polimerases dependentes de RNA estão em desenvolvimento.
Remdesivir (GS5734)
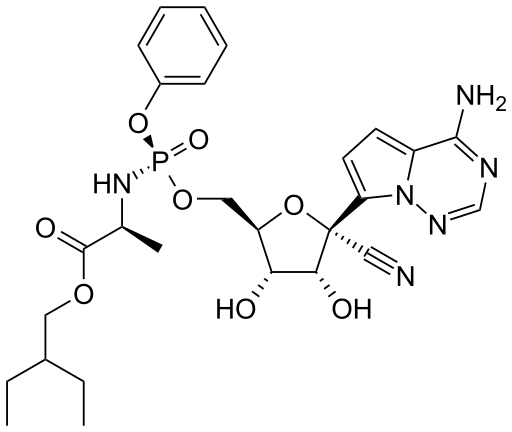
Uma droga antiga que foi testada contra o SARS-CoV-2 foi remdesivir. Esta foi originalmente desenvolvida como um tratamento contra a Hepatite C (um flavivírus), vírus Ebola e vírus Marburg (ambos filovírus), mas foi altamente ativa em estudos com células cultivadas contra o SARS-CoV-1, MERS CoV e SARS-CoV-2 (o agente causador da COVID-19). Por esta razão remdesivir é às vezes descrita como uma droga pan-viral, o que seria esperado, visto que todos os vírus de RNA devem codificar para suas próprias RNA polimerases.
Remdesivir é um análogo de adenosina cujo açúcar é ribose. Assim como o AZT, é uma pró-droga que tem que ser metabolizada a uma forma ativa (ou seja, fosforilada) antes de poder agir como um substrato terminalizador de cadeia para a RNA polimerase.
Como foi dito neste Capítulo 23 os Coronavírus são os maiores vírus de RNA conhecidos e o SARS-CoV-2 codifica para uma proteína, a NSP14 (ExoN), que age como uma exonuclease de revisão de leitura. Tal atividade seria um problema com o uso de uma droga terminalizadora de cadeia de nucleotídeos. Entretanto, embora tenha sido mostrado que um vírus da hepatite murina mutante que não tem o ExoN é mais sensível ao remdesivir, a droga inibe a polimerase (NSP12) mesmo na presença da atividade de revisão de leitura e qualquer resistência pode ser sobrepujada pelo aumento da concentração da droga. Entretanto, um estudo chines inicial no qual 158 pacientes foram tratados com remdesivir, e 79 receberam cuidados padrões com o placebo ao invés do tratamento, não houve diferença no tempo de recuperação entre os grupos. Menos de 14% dos que receberam remdesivir morreram, comparado com 13% daqueles que receberam o placebo. O estudo foi parado quando 11,6% dos pacientes tratados com remdesivir tiveram efeitos adversos comparados com 5,1% do grupo placebo.
Favipiravir (T-705, Avigan)
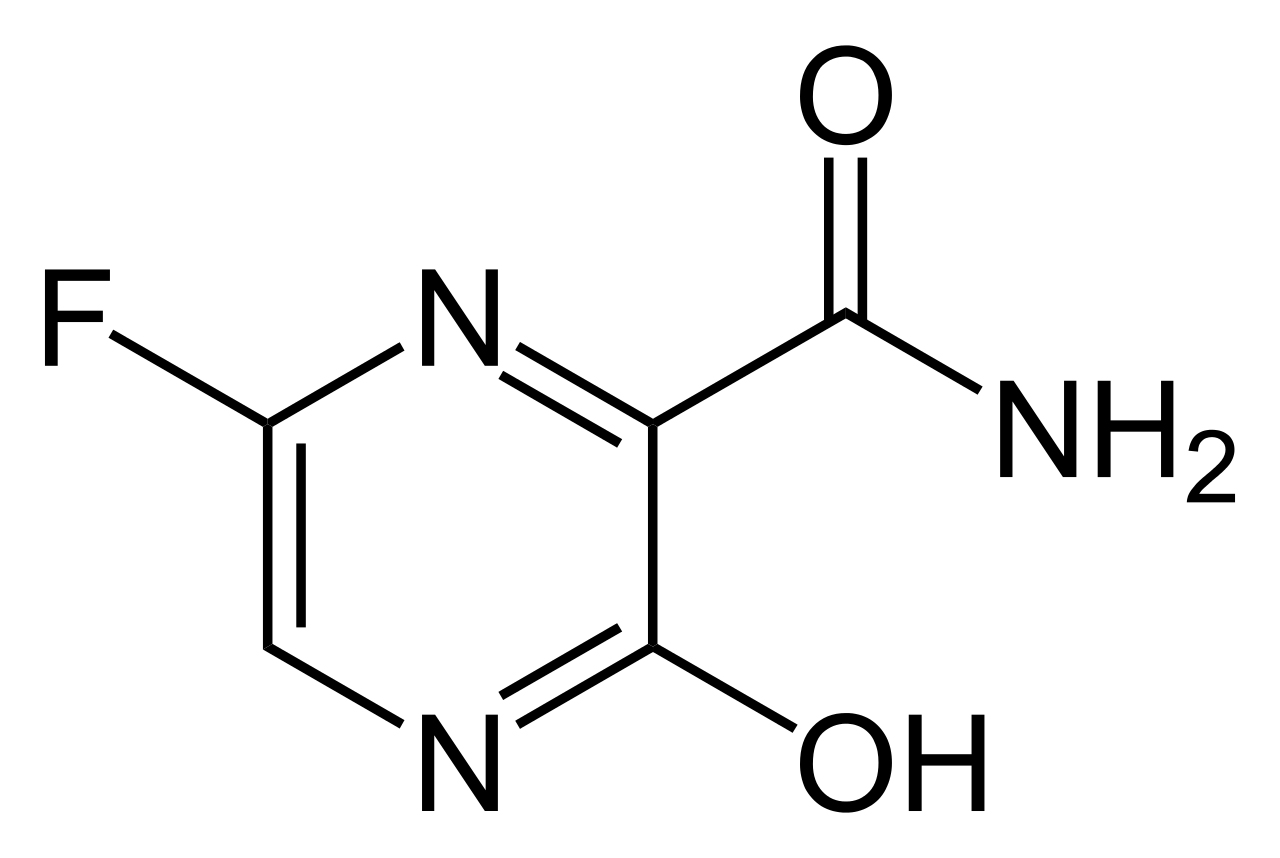
Favipiravir é um derivado de pirazinacarboxamida (6-Fluoro-3-hidroxi-2-pirazinacarboxamida) e outro inibidor da RNA polimerase dependente de RNA. É um análogo de purina que foi originalmente desenvolvido para tratar influenza, mas foi considerada de baixa eficácia. É uma pró-droga e sua molécula é muito mais simples do que a do remdesivir. É metabolizada a sua forma ativa, favipiravir-ribofuranosil-5'-trifosfato, pela hipoxantina guanina fosforibosiltransferase, que a converte à sua forma ribose-51-monofosfato, da qual é então convertida a trifosfato. Ela aparenta competir com GTP na síntese de RNA e leva a mutações letais no RNA viral não tendo efeito na síntese de RNA endógena.
Drogas lisossomotrópicas
Chloroquina e Hidroxicloroquina
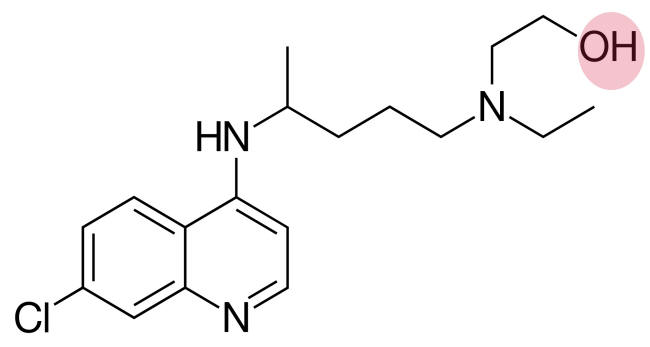
Cloroquina e hidroxicloroquina são largamente utilizadas como drogas anti-malária e também no tratamento de artrite reumatóide, lupus e porfiria cutânea tarda. Na primavera de 2020, uma publicação agora retirada sugeriu o uso da hidroxicloroquina em combinação com o antibiótico macrolídeo azitromicina como um tratamento para pacientes infectados pelo SARS-CoV-2. Experimentos em células Vero cultivadas mostraram que a hidroxicloroquina foi mais potente do que a cloroquina ao inibir o SARS-CoV-2 in vitro com um alto índice de seletividade; como resultado, muitos Estudos Clínicos sobre essas drogas sozinhas ou em combinação com outras drogas estão sendo conduzidos. Entretanto, tentativas anteriores de usá-las como anti-virais in vivo mostraram resultados contraditórios e às vezes elas exacerbaram a doença. Cloroquina inibiu a replicação do vírus Ebola, mas causou uma piora rápida em porcos-da-índia e não fez nenhuma diferença na mortalidade de ratos e hamsters. Similarmente, essas drogas pioraram os sintomas das infecções pelo vírus da chicungunha em macacos. Em abril de 2020, os primeiros resultados de um estudo com a hidroxicloroquina mostrou que em um grupo de veteranos americanos tratados contra a COVID-19, o risco de morte por qualquer causa foi significantemente maior naqueles que receberam a droga, comparados com aqueles que não receberam. Além disso, um Estudo Randomizado na China não mostrou evidências da eficácia da hidroxicloroquina.
Portanto, o que parece serem resultados promissores in vitro (isto é, cultura de células) não são frequentemente refletidos in vivo. Também se deve notar que enquanto essas drogas têm sido usadas clinicamente por muito tempo, especialmente como agentes anti-malária, elas não deixam de ter seus efeitos colaterais que incluem indução de arritmias cardíacas em pacientes geralmente predispostos, em quem as drogas bloqueiam os canais de íons que estão envolvidos no controle dos batimentos cardíacos. Azitromicina foi provavelmente usada no estudo original para controlar infecções bacterianas e ela pode aumentar as mortes cardiovasculares quando usados em associação com hidroxicloroquina em pacientes com artrite reumatóide.
Embora as cloroquinas (4-aminoquinolinas) sejam a muito
tempo usadas como anti-maláriais, seus mecanismos de ação são pouco
compreendidos. Elas são agentes lisossomotrópicos que inibem a acidificação dos
endossomos e lisossomos e podem bloquear a entrada de alguns vírus no citoplasma.
Isso tem sido sugerido pelos seus efeitos na replicação do HIV. No caso de
alguns coronavírus e outros vírus, um ambiente ácido é necessário para a mudança
conformacional na proteína de superfície do vírus (a proteína S no caso do
coronavírus) de forma que a região fusogênica desta molécula possa promover a
fusão da membrana do envelope viralcom a membrana endossomal/lisossomal
resultando na entrada do nucleocapsídeo no citoplasma. Outra possibilidade
sugerida pela modelagem molecular é que ambas as drogas possam se ligar ao ácido
siálico e gangliosídeos com alta afinidade. Um sítio de ligação a gangliosídeo
foi também identificado na proteína do SARS-CoV-2 S que pode estar envolvida no
acoplamento do vírus a intermediários lipídicos na membrana plasmática da célula
como um uma fase preliminar à ligação da proteína S ao receptor ACE-2. Assim, é
possível que as drogas ao ligar-se a gangliosídeos inibem a ligação do vírus aos
receptores da superfície da célula.