|
OBJECTIFS DU COURS
Comprendre le concept et l’importance de la tolérance
Connaître les facteurs qui déterminent l’induction de tolérance
Comprendre les mécanismes de l’induction de tolérance
Comprendre les concepts de l’auto-immunité et des maladies auto-immunes
Connaître les caractéristiques des maladies auto-immunes principales
Connaître les théories sur l’étiologie des maladies auto-immunes
|
TOLERANCE
INTRODUCTION
La notion de tolérance fait
référence à l’absence de réactivité immunologique spécifique envers un
antigène, faisant suite à une première rencontre avec ce même antigène.
Bien que la tolérance la plus importante soit la non-réactivité
vis-à-vis d’antigènes du soi, il est aussi possible d’induire une
tolérance à des antigènes du non soi. Lorsqu’un antigène induit de la
tolérance on dit qu’il est tolérogène.
TOLERANCE AUX ANTIGÈNES DU
SOI
Nous n'avons généralement pas
de réponse immunitaire forte contre nos propres (auto-) antigènes :
c’est un phénomène appelé tolérance au soi. Lorsque le système
immunitaire reconnaît un antigène du soi et répond fortement contre lui,
une maladie auto-immune peut alors se développer. Néanmoins, le système
immunitaire doit intrinsèquement reconnaître le CMH du soi pour répondre
contre un antigène étranger. Ainsi, le système immunitaire est
constamment mis au défi de distinguer le soi du non soi et déclencher
une réponse appropriée.
INDUCTION DE TOLERANCE AU
NON-SOI
La tolérance peut également
être induite contre des antigènes du non soi : cela peut se produire en
modifiant l'antigène, en l’injectant par des voies spécifiques telles
que par voie orale, ou encore lorsque l’antigène est administré alors
que le système immunitaire est en train de se développer par exemple.
Certaines bactéries et virus ont élaboré des moyens astucieux pour
induire une tolérance afin que l'hôte ne les tue pas. Ex: Les patients
de la lèpre de type lépromateuse ne déclenchent pas de réponse
immunitaire contre Mycobacterium leprae.
TOLERANCE AUX TISSUS ET
CELLULES
La tolérance à des antigènes tissulaires et cellulaires peut être
induite par l'injection de cellules souches hématopoïétiques chez des
animaux nouveau-nés ou sévèrement immunodéprimés (par irradiation létale
ou traitement pharmacologique). En outre, la greffe de moelle osseuse
allogénique ou de thymus en début de vie se traduit par une tolérance
aux cellules et tissus du donneur de greffe. Ces animaux sont connus
comme étant des chimères hématopoiétiques. Ces résultats ont des
applications pratiques importantes pour ce qui est des greffes de moelle
osseuse.
|
| |
TOLERANCE AUX ANTIGENES
SOLUBLES Un état de
tolérance vis-à-vis de nombreux antigènes T-dépendants et thymo-indépendants a
pu être obtenu dans différents modèles expérimentaux. Sur la base de ces
observations, il est clair qu'un certain nombre de facteurs déterminent si un
antigène va stimuler une réponse immunitaire ou induire la tolérance (Tableau
1).
|
Table 1
Facteurs déterminant l’induction de l’immunité ou de la tolérance suite
à une stimulation antigénique |
|
Facteurs
qui affectent la réponse à l’Ag |
En faveur de l’immunité |
En faveur de la tolérance |
|
Forme physique de l’antigène |
Molécules de grande taille, agrégées ou complexes. |
Molécules solubles, dépourvues
d’agrégats, plutôt petites, plutôt simples. Ag non apprêtés par les APC
ou apprêtées par des cellules n’exprimant pas le CMH II Ag |
|
Voie d’administration de l’Ag |
Sous-cutanée ou intramusculaire |
Orale ou parfois intraveineuse |
|
Dose d’antigène |
Dose optimale |
Très forte (ou parfois très faible) dose |
|
Age des animaux répondeurs |
Plus âgés et immunologiquement
matures |
Nouveau-nés (souris),
immunologiquement immatures |
|
Etat de différenciation
cellulaire |
Cellules complètement
différenciées; cellules T et B mémoires |
Relativement indifférenciées:
cellules B exprimant seulement des IgM (pas d’IgD), cellules T (ex :
cellules du cortex thymique) |
CARACTÉRISTIQUES
IMMUNOLOGIQUES DE LA TOLERANCE
La tolérance est différente de l'immunosuppression et de l'immunodéficience non
spécifique. Il s'agit d'un processus actif et antigène-dépendant induit en
réponse à l'antigène. Comme la réponse immunitaire, la tolérance est spécifique
et comme la mémoire immunologique, elle peut exister chez les lymphocytes T, les
B ou les deux, et enfin, comme la mémoire immunologique, la tolérance au niveau
des cellules T est plus durable que la tolérance au niveau des cellules B.
L'induction de la tolérance des lymphocytes T est plus facile à établir et
nécessite de relativement plus faibles quantités de tolérogène par comparaison
avec la tolérance des cellules B. Le maintien de la tolérance immunologique
exige la persistance de l'antigène. La tolérance peut être rompue naturellement
(comme dans les maladies auto-immunes) ou artificiellement (comme par exemple
sur les animaux de laboratoire, par irradiation aux rayons X, par traitements
médicamenteux ou par exposition à des antigènes donnant des réactivités croisées).
La tolérance peut être induite contre l’ensemble ou seulement contre certains
épitopes d’un antigène donné et la tolérance à un antigène unique peut exister
au niveau des lymphocytes B ou des lymphocytes T niveau ou aux deux niveaux.
|
| |
MECANISMES DE
L’INDUCTION DE TOLERANCE
Les mécanismes exacts de
l'induction et du maintien de la tolérance ne sont pas entièrement
compris. Les données expérimentales, cependant, indiquent plusieurs
possibilités.
Délétion
clonale
Les lymphocytes T et B sont confrontés à des auto-antigènes au cours
de leur développement et ces cellules subissent alors la délétion
clonale par un processus appelé apoptose ou mort cellulaire
programmée. Par exemple, les cellules T qui se développent dans le
thymus n’expriment ni CD4 ni CD8 au début du développement. Ces
cellules acquièrent ensuite l’expression de CD4 et de CD8 et sont
alors appelées cellules doubles-positives. Ces cellules expriment
des niveaux faibles de TCR αβ. Elles subissent une sélection
positive après interaction avec les molécules du CMH de classe ou de
classe II exprimées sur l'épithélium cortical du thymus. Au cours de
ce processus, les cellules ayant une faible affinité pour le CMH
sont sélectionnés positivement. Les cellules non sélectionnées
meurent par apoptose, un processus appelé «mort par négligence».
Ensuite, les cellules perdent soit CD4 ou CD8. Ces cellules T
rencontrent alors des peptides du soi présentés par les molécules du
CMH du soi exprimées sur les cellules dendritiques. Les cellules T
possédant des récepteurs de haute affinité pour le CMH + peptide du
soi subissent alors une délétion clonale aussi appelée sélection
négative par induction d'apoptose. Toute perturbation de ce
processus peut conduire à l'évasion de cellules T auto-réactives qui
peuvent alors déclencher des maladies auto-immunes. De même, les
cellules B subissent la délétion clonale lorsqu’elles rencontrent
des auto-antigènes associés aux cellules ou solubles au début de
leur différenciation. Ainsi, la délétion clonale joue un rôle clé
pour assurer la tolérance vis-à-vis des auto-antigènes.
Tolérance périphérique
La délétion clonale n'est pas un système infaillible et souvent les
cellules T et B ne subissent pas la délétion et donc ces cellules
peuvent potentiellement être responsables d’une maladie auto-immune
après avoir atteint les organes lymphoïdes périphériques. De ce
fait, le système immunitaire a mis au point plusieurs points de
contrôle supplémentaires de sorte que la tolérance puisse être
maintenue.
Mort
cellulaire induite par l’activation
Lors de l'activation, les lymphocytes T produisent non seulement des
cytokines ou exercent leurs fonctions effectrices, mais meurent
aussi par mort cellulaire programmée ou apoptose. Dans ce processus,
le récepteur de mort (Fas) et son ligand (FasL) jouent un rôle
crucial. Ainsi, les cellules T normales expriment Fas mais pas FasL.
Lors de l'activation, les lymphocytes T se mettent à exprimer FasL
qui oeut alors se lier à Fas ce qui déclenche l'apoptose par
activation de la caspase-8. L'importance de Fas et FasL est
clairement démontrée par l'observation que des souris présentant des
mutations dans Fas (mutation lpr) ou FasL (mutation gld) développent
un syndrome lymphoprolifératif et des maladies auto-immunes sévères
et meurent dans les 6 mois, alors que les souris normales vivent
jusqu'à 2 ans. Des mutations similaires de ces gènes apoptotiques
chez l’homme conduisent à une maladie appelée le syndrome auto-immun
lymphoprolifératif.
Anergie
clonale
Les cellules T auto-réactives deviennent anergiques (non répondeuses)
à l’antigène lorsqu'elles sont exposées à des antigènes présentés
par des cellules présentatrices d'antigènes (APC) n’exprimant pas
les molécules de co-stimulation CD80 (B7-1) ou CD86 (B7-2). En outre,
tandis que l'activation des cellules T par l'intermédiaire de la
molécule CD28 entraîne la production d'IL-2, l'activation de la
molécule CTLA-4 conduit à l'inhibition de la production d'IL-2 et à
l'anergie. Finalement, les cellules B diminuent l’expression de leur
IgM de surface et deviennent anergiques lorsqu'elles sont exposées à
de fortes quantités d'antigènes solubles. Ces cellules peuvent aussi
augmenter l’ expression des molécules Fas à leur surface.
L’interaction de ces cellules B avec des cellules T exprimant FasL
entraîne leur mort par apoptose.
L’ignorance
clonale
Les lymphocytes T réactifs vis-à-vis d’un auto-antigène absent du
thymus vont maturer normalement et migrer vers la périphérie, mais
ils ne vont jamais rencontrer l'antigène approprié, car son
expression est restreinte à des tissus inaccessibles. Ces cellules
peuvent mourir par manque de stimulation. Les cellules B auto-réactives,
ayant échappé à la délétion, peuvent ne pas trouver l'antigène ou ne
pas bénéficier de l'aide spécifique des cellules T et ne vont donc
pas être activées et finiront par mourir.
Anticorps
anti-idiotypiques
Ce sont des anticorps qui sont produits contre des idiotypes
spécifiques d'autres anticorps. Les anticorps anti-idiotypiques sont
produits au cours du processus de tolérisation et ont été mis en
évidence dans des modèles d’animaux tolérants. Ces anticorps peuvent
empêcher le récepteur des cellules B d'interagir avec l'antigène.
Cellules T
régulatrices (anciennement appelées cellules T suppressives)
Récemment, une population distincte de cellules T appelées cellules
T régulatrices a été découverte. Les cellules T régulatrices sont
très diverses mais la population la mieux caractérisée exprime les
marqueurs CD4 et CD25. Etant donné que les cellules T CD4
conventionnelles activées expriment aussi CD25, il était difficile
de distinguer les cellules T régulatrices des lymphocytes T activés.
Les dernières études montrent que les cellules T régulatrices
peuvent êtret définies par l'expression du facteur de transcription
Foxp3 de la famille « forkhead ». L’expression de Foxp3 est
nécessaire pour le développement et la fonction des cellules T
régulatrices. Le mécanisme précis par lequel les cellules T
régulatrices suppriment d’autres fonctions des cellules T n'est pas
clair. L'un des mécanismes met en jeu la production de cytokines
immunosuppressives telles que le TGF-β et l'IL-10. Les mutations
génétiques de Foxp3 chez l'homme conduisent au développement d'une
maladie auto-immune grave et rapidement mortelle connue sous le nom
de syndrome IPEX pour Immune dysregulation, Polyendocrinopathy,
Enteropathy, X-linked. Cette maladie fournit la preuve la plus
évidente que les cellules T régulatrices jouent un rôle essentiel
dans la prévention des maladies auto-immunes.
Rupture de la
tolérance
La tolérance induite expérimentalement peut être rompue par
l'absence prolongée de l'exposition à l’agent tolérogène, par des
traitements qui endommagent sévèrement le système immunitaire
(x-irradiation) ou par la vaccination avec des antigènes donnant
lieu à des réactions croisées. Ces observations sont importantes
pour la compréhension des maladies auto-immunes.
|
| |
AUTOIMMUNITE
DEFINITION
L'auto-immunité peut être définie comme la rupture
des mécanismes responsables de la tolérance et l’induction d'une réponse
immunitaire contre les composants du soi. Une telle réponse immunitaire
n’a pas toujours des conséquences nuisibles (par exemple, les anticorps
anti-idiotypiques). Cependant, dans de nombreuses maladies auto-immunes,
il est reconnu que les produits de la réponse immunitaire vont causer
des dommages au soi.
MECANISMES
EFFECTEURS DES MALADIES AUTOIMMUNES
Aussi bien les anticorps que
les cellules T effectrices peuvent être impliquées dans les dommages
causés lors des maladies auto-immunes.
CLASSIFICATION
GENERALE
Les maladies auto-immunes sont
généralement classées sur la base de l'organe ou du tissu concerné. Ces
maladies peuvent être soit spécifique d'un organe donné et, dans ce cas,
la réponse immunitaire est dirigée contre un antigène (s) associé à
l'organe cible endommagé ou soit non spécifique d’organe : dans ce cas,
l'anticorps est dirigé contre un antigène non particulièrement associé à
l'organe cible (Tableau 2). L'antigène impliqué dans la plupart des
maladies auto-immunes peut souvent être déduit du nom de la maladie
(Tableau 2).
PREDISPOSITIONS
GENETIQUES A L’AUTOIMMUNITE
Des études chez la souris
ainsi que des observations chez l'homme suggèrent l’existence d’une
prédisposition génétique pour les maladies auto-immunes. L’association
entre certains allèles HLA et maladies auto-immunes a été notée (HLA:
B8, B27, DR2, DR3, DR4, DR5, etc...)
|
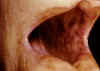 Figure 1 Hyperpigmentation de la muqueuse buccale dans la maladie
d’Addison © Bristol Biomedical Archive. Reproduit avec
permission
Figure 1 Hyperpigmentation de la muqueuse buccale dans la maladie
d’Addison © Bristol Biomedical Archive. Reproduit avec
permission
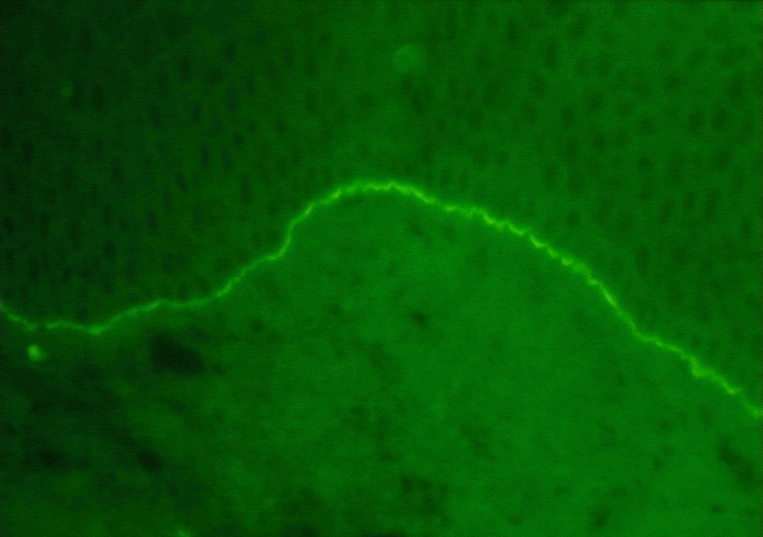
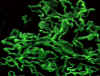 Figure 2 Marquage par immunofluorescence des immunoglobulines G (IgG)
montrant un profil linéaire et lisse dans le syndrome de Goodpasture
© Bristol Biomedical Archive. Reproduit avec permission
Figure 2 Marquage par immunofluorescence des immunoglobulines G (IgG)
montrant un profil linéaire et lisse dans le syndrome de Goodpasture
© Bristol Biomedical Archive. Reproduit avec permission
 Figure 3 Immunofluorescence de pemphigus vulgaris ©
Bristol Biomedical Archive. Reproduit avec permission
Figure 3 Immunofluorescence de pemphigus vulgaris ©
Bristol Biomedical Archive. Reproduit avec permission
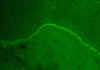 Figure 4 Membrane muqueuse pemphigoïde - immunofluorescence
© Bristol Biomedical Archive. Reproduit avec permission
Figure 4 Membrane muqueuse pemphigoïde - immunofluorescence
© Bristol Biomedical Archive. Reproduit avec permission
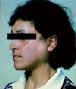 Figure 5 Elargissement de la parotide dans le syndrome de Sjogren
© Bristol Biomedical Archive. Reproduit avec permission
Figure 5 Elargissement de la parotide dans le syndrome de Sjogren
© Bristol Biomedical Archive. Reproduit avec permission
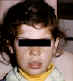
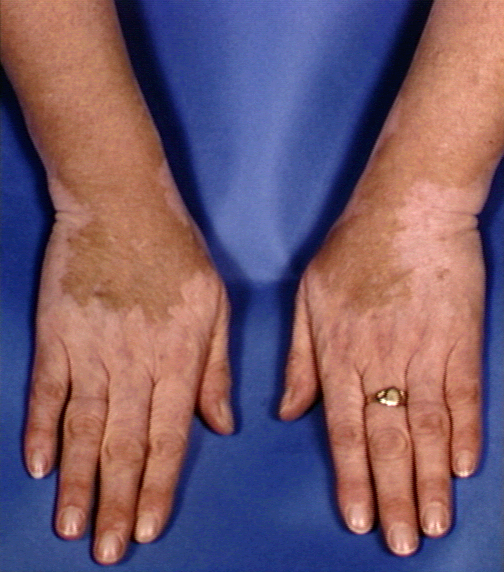
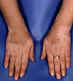 Figure 6 Dépigmentation dans le vitiligo © Bristol
Biomedical Archive. Reproduit avec permission
Figure 6 Dépigmentation dans le vitiligo © Bristol
Biomedical Archive. Reproduit avec permission
|
ETIOLOGIE DES MALADIES
AUTO-IMMUNES
L'étiologie exacte des maladies
auto-immunes n'est pas connue. Cependant, diverses théories ont été proposées.
Il s'agit notamment des théories de l'antigène séquestré, de l'échappement de
clones auto-réactifs, de la perte de cellules suppressives, et celle des
réactions croisées entre des antigènes exogènes (agents pathogènes) et des auto-antigènes
modifiés (infections virales et modifications chimiques).
Antigène séquestré
Les cellules lymphoïdes peuvent ne pas être exposés à des antigènes du soi
au cours de leur différenciation, car il se peut que ces antigènes
apparaissent tardivement dans le développement ou que leur expression soit
confinée à des organes spécialisés (par exemple, les testicules, le cerveau,
les yeux, etc.) Une libération d'antigènes à partir de ces organes,
résultant d'une lésion ou une chirurgie traumatique accidentelle peut
résulter en la stimulation d'une réponse immunitaire et le déclenchement
d'une maladie auto-immune.
Echappement de clones
auto-réactifs
La sélection négative dans le thymus peut ne pas être pleinement
fonctionnelle pour éliminer les cellules auto-réactives. En effet, tous les
antigènes du soi ne peuvent être présents dans le thymus et certains
antigènes peuvent ne pas être correctement présentés dans le thymus.
Défaut des cellules T
régulatrices
Il y a souvent moins de cellules T régulatrices (ou des cellules T
régulatrices moins fonctionnelles) dans beaucoup de maladies auto-immunes.
|
Table 2
Spectre des maladies auto-immunes, organes ciblés et tests diagnostiques |
|
|
Maladie |
Organe |
Anticorps
contre |
Diagnostique |
|
Spécifiques d’organes

Non-Spécifiques
d’organes
|
Thyroïdite de Hashimoto'
|
Thyroïde |
Thyroglobuline, Peroxydase thyroïdienne (microsomale) |
RIA, Passive, CF,
hémagglutination
|
|
Myxoedème primaire |
Thyroïde |
Récepteur cytoplasmique au TSH
|
Immunofluorescence
(IF) |
|
Maladie de Graves |
Thyroïde |
|
Bio-essai, compétition avec le
récepteur au TSH
|
|
Anémie pernicieuse |
Globules rouges |
Facteur intrinsèque (IF), cellules pariétales
gastriques |
Liaison B-12/IF en immunofluorescence
|
Maladie d’Addison
(Fig 1)
|
Surrénales |
Cellules surrénales |
Immunofluorescence |
|
Ménopause prématurée
|
Ovaires |
Cellules productrices de stéroïdes |
Immunofluorescence |
|
Infertilité masculine |
Sperme |
Spermatozoïdes |
Agglutination, Immunofluorescence
|
|
Diabète juvénile
insulino-dépendant
|
Pancréas |
Cellules beta des ilots pancréatiques |
|
|
Diabète insulino-résistant
|
Systémique |
Récepteur à l’insuline |
Compétition pour le récepteur |
|
Allergie atopique |
Systémique |
Récepteur beta-adrénergique
|
Compétition pour le récepteur |
| Myasthenia graves |
Muscle |
Muscle, récepteur à
l’acétylcholine
|
Immunofluorescence, compétition
pour le récepteur
|
|
Syndrome de Goodpasture
|
Reins, poumons
|
Membranes basales des reins et poumons |
Immunofluorescence (marquage linéaire)(Fig. 2) |
| Pemphigus |
Peau |
Desmosomes |
Immunofluorescence
(Fig 3) |
|
Pemphigoïde bulleuse |
Peau |
Membranes basales de la peau
|
Immunofluorescence
(Fig 4) |
|
Uvéite phacogénique |
Lentille |
Protéine de lentille |
|
|
Anémies
hémolytiques |
Globules rouges, Plaquettes |
Globules rouges |
Hémagglutination passive
Test direct de Coomb
|
|
Thrombocytopénie idiopathique
|
|
Plaquettes |
Immunofluorescence |
|
Cirrhose biliaire primaire
|
Foie |
Mitochondrie |
Immunofluorescence |
|
Neutropénie idiopathique
|
Neutrophiles |
Neutrophiles |
Immunofluorescence |
|
Colite ulcéreuse |
Colon |
Lipopolysaccharide du colon
|
Immunofluorescence |
|
Syndrome de Sjogren |
Glandes sécrétoires
(Fig 5)
|
Conduit de mitochondries |
Immunofluorescence |
| Vitiligo |
Peau, articulations
|
Mélanocytes (fig 6) |
Immunofluorescence |
|
Arthrite rhumatoïde |
Peau, reins, articulations etc… |
IgG |
Agglutination de billes de latex-IgG |
|
Lupus érythémateux disséminé
|
Articulations, etc… |
ADN, ARN, nucléoprotéines
|
Agglutination de billes de
latex-ADN ou -ARN, IF (marquage granulaire dans le rein)
|
|
|
|
|
|
|
Les
maladies sont listées depuis les plus spécifiques d’organes (haut) vers les
moins spécifiques (bas) |
Antigènes donnant des
réactions croisées
Les antigènes de certains agents pathogènes peuvent avoir des déterminants
qui réagissent de façon croisée avec les antigènes du soi et une réponse
immunitaire contre ces déterminants peut conduire à des cellules effectrices
ou des anticorps contre les antigènes tissulaires. La néphrite postérieure à
l’infection à streptocoques, la cardite et la présence d’anticorps
anticardiolipine au cours de la syphilis et l'association entre l’infection
à Klebsiella et la spondylarthrite ankylosante sont des exemples de telles
réactivités croisées.
DIAGNOSTIC
Le diagnostic de maladies auto-immunes
est basé sur les symptômes et la détection d'anticorps (et/ou de lymphocytes T)
réactifs contre des antigènes des tissus et des cellules impliquées dans la
pathologie. Les anticorps dirigés contre des antigènes associés à la cellule/tissu
sont détectés par immunofluorescence. Les anticorps dirigés contre des antigènes
solubles sont normalement détectés par ELISA ou des tests radio-immunologiques (voir
le tableau ci-dessus). Dans certains cas, un dosage biologique / biochimique
peut être utilisé (par exemple : maladie de Basedow, anémie pernicieuse).
TRAITEMENT
Les objectifs du traitement des
maladies auto-immunes sont de réduire les symptômes et de contrôler la réponse
auto-immune tout en maintenant la capacité de l'organisme à combattre les
infections. Les traitements varient beaucoup et dépendent de la maladie et de
ses symptômes spécifiques: l’utilisation d’anti-inflammatoires (corticoïdes) et
de traitements immunosuppressifs (comme le cyclophosphamide, l’azathioprine, la
cyclosporine) est la méthode actuelle de traitement de la plupart des maladies
auto-immunes. Des recherches poussées sont menées pour développer des
traitements innovants qui incluent: la thérapie anti-TNF alpha dans l'arthrite,
l'administration de l'antigène par voie orale pour déclencher la tolérance,
d’anticorps anti-idiotypiques, de peptides antigéniques, d’anticorps anti-récepteurs
dirigés contre l'IL-2, d’anticorps anti-CD4, anti-TCR etc…
MODELES DE MALADIES
AUTOIMMUNES
Il y a un certain nombre de modèles
animaux expérimentaux et naturels pour l'étude des maladies auto-immunes. Les
modèles expérimentaux incluent l’encéphalite auto-immune expérimentale, la
thyroïdite expérimentale, l'arthrite induite par adjuvant, etc…
Des modèles naturels de maladies
auto-immunes sont l'anémie hémolytique chez les souris NZB, le lupus
érythémateux disséminé chez les souris NZB / NZB (BW), les souris BXSB et LMR et
le diabète chez les souris obèses.
|

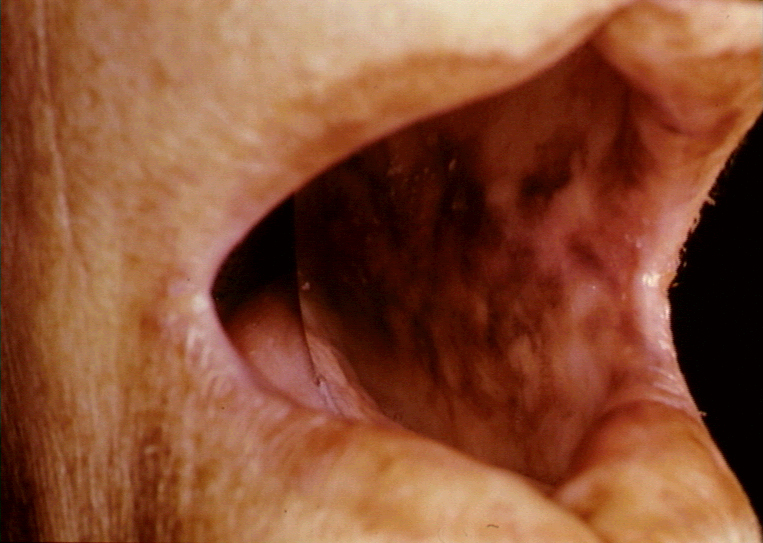 Figure 1 Hyperpigmentation de la muqueuse buccale dans la maladie
d’Addison © Bristol Biomedical Archive. Reproduit avec
permission
Figure 1 Hyperpigmentation de la muqueuse buccale dans la maladie
d’Addison © Bristol Biomedical Archive. Reproduit avec
permission