|
|
 |
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Dr Richard Hunt |
BACTERIOLOGÍA | INMUNOLOGÍA | MICOLOGÍA | PARASITOLOGÍA | VIROLOGÍA | ||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
VIDEOCONFERENCIA |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Lectura: Murray et al Capítulo 16 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
OBJETIVOS
Elucidar los fármacos que son usados en la actualidad como agentes
antivirales y determinar el porqué son efectivos. El mecanismo de acción
de estos fármacos también será discutido |

|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
VEA TAMBIÉN |
Los fármacos anti-bacteriales como los antibióticos tipo penicilina han probado ser muy eficaces puesto que actúan contra una estructura bacteriana, la pared celular, que no se encuentra en las células eucarióticas. Al contrario, la mayoría de los agentes anti-virales han probado tener poca eficacia terapéutica dado que el virus usa reacciones metabólicas del huésped y por tanto, muchos de los agentes anti-virales también son anti-celulares. Por ello, el abordaje alternativo de estimular las respuestas inmunes del huésped mediante vacunas ha sido el más usado. No obstante, hay reacciones (i.e. enzimáticas) que son codificadas por le virus y por tanto ofrecen un potencial blanco específico para virus. Este es el caso particular de los virus que tienen genomas grandes y codifican sus propias enzimas de replicación. Aun así, desafortunadamente, muchos anti-virales que aparentemente son eficaces in vitro no lo son in vivo.
Un fármaco
anti-viral eficaz debe de: Un fármaco ideal debe de ser:
Un fármaco ideal NO debe de ser:
La toxicidad de una droga anti-viral puede ser aceptable cuando no se tiene otra alternativa: como, por ejemplo, en rabia sintomática o fiebre hemorrágica Obviamente, un buen fármaco debe mostrar mucha más toxicidad hacia el virus que hacia la célula huésped. Se mide su selectividad mediante el índice terapéutico del fármaco
Índice
terapéutico (I.T.): Dosis mínima que es tóxica a la célula Los fármacos efectivos tienen un I. T. de 100-1000 o mejor. Al igual que con los anti-bacterianos, se encuentra en “talón de Aquiles” del virus. Este puede ser alguna enzima única del virus de manera que el fármaco no sea tóxico para la célula huésped. La siguiente lista es de los virus conocidos por codificar sus propias enzimas. Entre dichas enzimas: proteasas, enzimas chapadoras de ARNm, neuraminidasas, ribonucleasas, kinasas y enzimas que quitan las envolturas.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Las ventanas emergentes de la Estructura Molecular muestran las estructuras químicas y tridimensionales
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
El primer fármaco antiviral aprobado fue la idoxuridina (1963), un análogo de pirimidina que inhibe la síntesis viral de AND. Aún se emplea de modo tópico en las queratitis herpéticas epiteliales pero ha sido en su mayoría remplazado por otras drogas menos tóxicas. Es tóxica porque carece de especificidad, i.e. el fármaco inhibe la polimerización de AND del huésped así como la del virus. Una de las mejores drogas anti-virales que tenemos data de 1983: Aciclovir (acicloguanosina) que es un análogo de las purinas. Inhibe la replicación del ADN del herpes. También es un análogo de nucleósido, en contraste con la idoxiuridina, es altamente específico y no exhibe efectos adversos tóxicos severos… para la razón de esto, vea más adelante.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
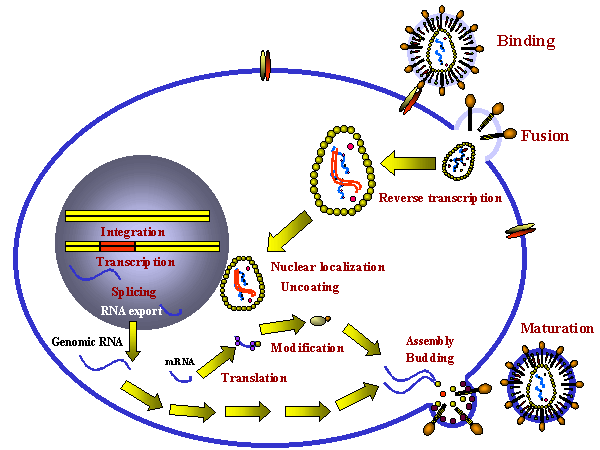 Figura 1
Figura 1Blancos celulares de los fármacos |
PROBABLES ETAPAS DEL CICLO VITAL EN LAS QUE UN ATAQUE ANTI-VIRAL PUEDE INICIARSE El ciclo vital de un virus implica varias etapas como el de unión a la superficie celular, replicación, síntesis proteica etc. y todas estas etapas pueden ser blancos de los fármacos antivirales. Entre las etapas del ciclo vital que han sido objetivos de agentes terapéuticos potenciales tenemos:
Haremos una revisión de cada una de estas etapas del ciclo vital (figura 1) en las siguientes secciones.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
FUENTES EN LA RED (en inglés) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
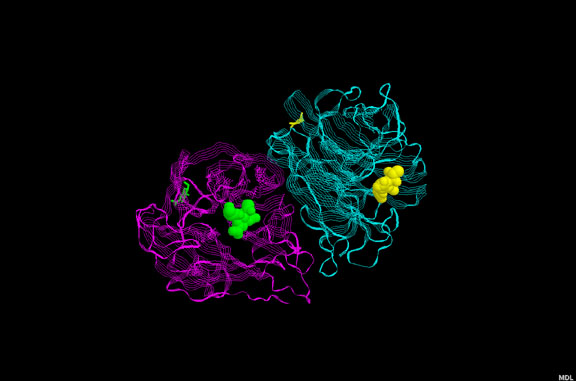
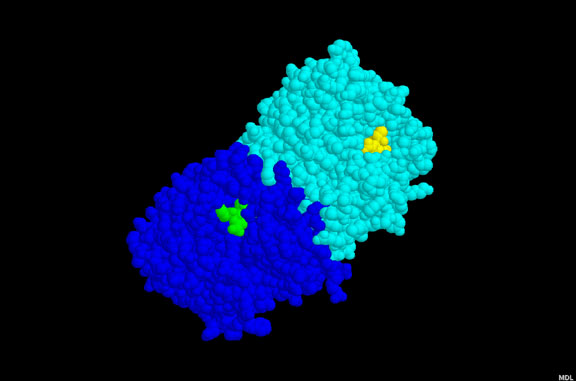
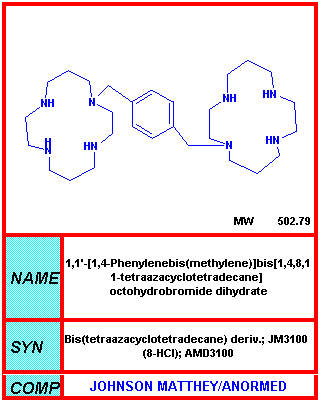 Figura 1a
Figura 1aAMD3100 |
UNIÓN AL RECEPTOR O ABSORCIÓN EN VESÍCULAS INTRACELULARES Aquí, hasta hace poco, no había ningún fármaco bueno que impidiera la unión del receptor a ningún virus (pero refiérase a los inhibidores de sialidasa de influenza más adelante). Sin embargo, entre las posibilidades se incluye el uso de un péptido que imite el receptor tal como la proteína soluble CD4. Este se uniría a la gp120 del VIH e impediría su unión al receptor de superficie celular. No obstante, hay un problema de estabilidad. La proteína soluble es rápidamente metabolizada y depurada de la circulación, i.e. una concentración eficaz no se logra por un periodo significativo. Se han hecho muchos intentos para estabilizar las proteínas pero con muy poco éxito. Se han dado intentos de acoplar las CD4 solubles a toxinas para eliminar células infectadas, pero tampoco han sido eficaces. En algunos casos, las CD4 solubles pueden hacer que el virus sea más infeccioso en estudios de laboratorio. Se desconoce la razón de que esto ocurre pero una posible explicación puede ser que la unión a la gp120 causa un cambio conformacional en esta última dándole mayor afinidad para el co-receptor que es importante, junto con el antígeno CD4, en la infección de una célula por el VIH (vea el capítulo de VIH, sección 7). También es posible que la CD4 soluble unida a la gp120 sea promotora de la fusión. El PRO 542 es una forma tetramérica del antígeno CD4 soluble genéticamente fusionado a una inmunoglobulina para mayor estabilidad. Esta proteína CD4-inmunoglobulina comprende los dominios D1 y D2 del Cd4 humano y las regiones constantes de cadenas pesada y ligera de la IgG2 humana. Tiene una alta afinidad para la gp120. Para que el VIH infecte una célula, debe de unirse tanto al antígeno CD4 como a un co-receptor, un receptor de quimiokinas. Los receptores de quimiokinas se unen a quimiokinas y pueden bloquear la unión del VIH a la gp120. Algunos derivados de estas quimiokinas (RANTES) han sido usados como agentes para bloquear la unión viral. Además de unirse con el receptor CCR5 de quimiokinas, estos derivados, al igual que las quimiokinas naturales, provocan una regulación a la baja del co-receptor mediante endocitosis, haciendo más difícil que el virus se una. Las quimiokinas como el RANTES son pro-inflamatorias y quimiotácticas para los leucocitos, sin embargo estas propiedades pueden ser aminoradas mediante modificaciones químicas en la porción N-terminal de la molécula. Estos derivados de las quimiokinas con excelentes antagonistas de unión del VIH y han demostrado que protegen durante la exposición vaginal al mismo en monos. Los anticuerpos monoclonales anti-correceptores también se están desarrollando para el bloqueo de la unión del virus. Otro abordaje en estudio es el uso de péptidos análogos a la secuencia transmembranal del correceptor; estos impiden la interacción entre las siente alfa-hélices trasmembranales del correceptor. AMD-3100
AgentES QUE BLOQUEAN LA FUSIÓN DEL VIH CON LA CÉLULA HUÉSPED AL INTERACTUAR CON gp41 Enfuvirtide
Los péptidos derivados de la gp41 pueden inhibir la infección, probablemente al bloquear la interacción de la gp41 con las proteínas de membrana celular durante la fusión o al detener el cambio conformacional que resulta de la asociación de dos moléculas gp41, que es necesario para la fusión. El enfuvirtide (Fuzeon) es un péptido de 36 aminoácidos que corresponde a los residuos 127-162 de la gp41 e impide el cambio conformacional. En ensayos clínicos, se ha logrado una reducción de casi dos medidas logarítmicas en los niveles plasmáticos del virus. Este fármaco fue aprobado en el 2003 pero hay reportes recientes que sugieren que tiene una biodisponibilidad baja y el surgimiento de variantes resistentes. Hay una hendidura en la gp41 que puede albergar alguna molécula inhibidora pequeña. Se han identificado péptidos con D-amino ácidos que podrían entrar en esta hendidura e inhibir la fusión. OTROS
RFI-641 Nombre químico: 2H-Imidazo (4,5-c) piridin-2-ona, 1-ciclopropil-1,3-dihidro-3-((1-(3-hidroxipropil)-1H-benzimidazol-2-il) metil)- BMS-433771 es un inhibidor de fusión de VSR. Actúa mediante inhibición de la fusión de membrana inducida por la proteína F viral y es activa contra amas cepas, A y B, del VSR. Es eficaz contra la infección por el VSR en dos modelos de roedores que fueron dosificados oralmente previo a la infección y puede tener uso clínico.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
PÉRDIDA DE LA ENVOLTURA VIRAL La pérdida de la envoltura del virus (i.e. la pérdida de la envoltura lipídica de los virus envueltos o la pérdida de las proteínas de la nucleocápside en los virus no envueltos) usualmente se da en endosomas a bajo pH o en lisosomas, como resultado de un fusógeno pH-dependiente. Nota: Algunos virus no necesitan de un ambiente acídico para la fusión y se fusionan con la membrana plasmática; este es el caso de los herpesvirus y del VIH y conlleva la formación de sincitios. |
||||||||||||||||||||||||||||||||||||||||||||||||||||
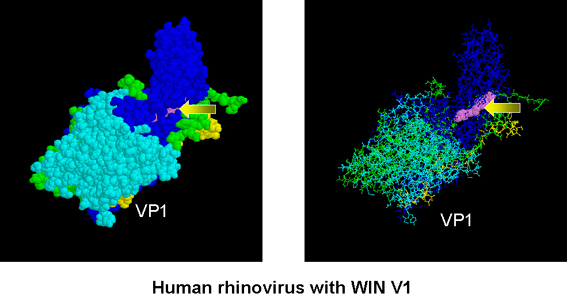 Rinovirus humano con un WIN V1 (flechas) incrustadas en una hendidura en
la proteína VP1
Rinovirus humano con un WIN V1 (flechas) incrustadas en una hendidura en
la proteína VP1
|
La
arildona y los compuestos WIN
Pleconaril
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Figura 4 |
Amantadina
Rimantadina
Estos fármacos son buenos como profilaxis oral contra la influenza A (pero no para la influenza B). Son una buena alternativa a la vacuna en pacientes inmunocomprometidos y en mayores de edad. Además de esto, no tienen otra utilidad en países occidentales. La rimantadina como profilaxis ha sido usada ampliamente en países de la antigua Unión de Repúblicas Socialistas Soviéticas (URSS). Ambos fármacos están aprobados para su uso en EEUU. El interés en estas drogas ha resurgido puesta la posibilidad de una pandemia de gripe aviar dado que actualmente no hay vacuna para este tipo del virus de la influenza (H5N1) y tomaría varios meses el desarrollarla luego de que se identifique la cepa pandémica. En la temporada de influenza del 2005-2006, el 92% de las cepas H3N2 examinadas tenían mutaciones que les conferían resistencia a este tipo de fármacos, igual ocurrió con el 25% de las cepas H1N1 evaluadas. Problemas similares se observaron en el 2006-2007 y por ello estos fármacos no son recomendados hasta que el porcentaje de resistencias en las cepas circulantes más importantes se reduzca.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
FUENTES EN
LA RED (en inglés) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
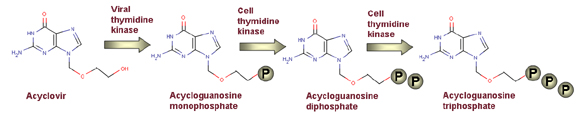
El aciclovir es fosforilado primero por una cinasa viral a acicloGMP y luego por cinasas celulares a acicloGDP y acicloGTP Figura 5 |
SÍNTESIS DE ÁCIDOS NUCLÉICOS Los mejores fármacos anti-virales con los que contamos son de este tipo. Estos son selectivos puesto que:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
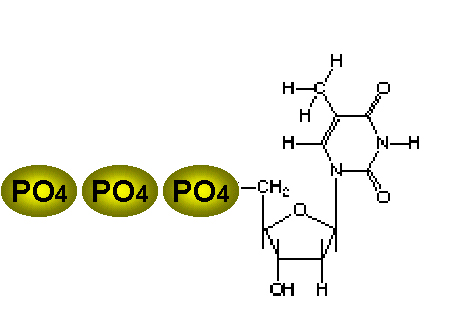 Tres fosfatos son añadidos a la timidina. El primero es agregado por la
enzima viral y el esto por enzimas celulares
Tres fosfatos son añadidos a la timidina. El primero es agregado por la
enzima viral y el esto por enzimas celularesFigura 6 |
Substratos de timidina cinasa (o timidina kinasa) La timidina cinasa (figura 6) del virus herpes simplex (y de otros virus) le permite al mismo crecer en células que no tienen una alta concentración de precursores fosforilados de ácidos nucleicos. Estas usualmente son células que no están replicando su genoma (i.e. células nerviosas). Las células en reposo, sin embargo, tienen nucleósidos no fosforilados. Al portar su propia cinasa, el virus puede crecer en células que no están en división al fosforilar los nucleósidos de las mismas. El nombre de la enzima es un término equivocado dado que sólo puede trabajar en otros nucleósidos diferentes de la timidina (aun cuando resulta que la timidina es el mejor sustrato), i.e. la enzima no es sustrato - específica. Esto es contrario a la timidina kinasa celular que es muy específica para la timidina porque la célula tiene otras enzimas para la fosforilación de otros nucleósidos. Esta falta de especificidad de la enzima viral le permite trabajar en los fármacos análogos de los nucleósidos y fosforilarlos. La enzima celular, por su alta especificidad, es mucho menos propensa a esto (y generalmente no fosforila en lo absoluto el fármaco). El hecho de que la enzima viral pueda fosforilar en fármaco tiene otra ventaja. Se puede administrar el análogo de nucleósido en una forma no fosforilada. Esto es de utilidad puesto que es difícil que un fármaco fosforilado penetre la célula porque las membranas plasmáticas son poco permeables a los compuestos fosforilados en la ausencia de proteínas de transporte específicas. Aun así, la necesidad de activación restringe el uso del fármaco a virus que tengan su propia timidina cinasa o que causen que la célula sobre produzca su enzima endógena (lo que, si se tiene suerte, puede activar el fármaco aunque en mucho menor medida). Para recapitular, la gran ventaja de estos fármacos esta dada porque:
La mayor parte de los fármacos inhibidores de la síntesis nucleica son análogos de nucleósidos con un sacárido alterado, una base o ambas. El aciclovir (acicloguanosina) es el mejor ejemplo de tales fármacos y es usado para tratar infecciones de herpesvirus. Penetra en la célula a través de la membrana plasmática en su forma de nucleósido y luego es específicamente fosforilado, dentro de la célula por la timidina cinasa del herpesvirus, a una forma más activa. Este luego bloquea la síntesis de ADN al inhibir la polimerización; es un finalizador de cadenas.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
 Aciclovir
AciclovirFigura 7 |
INHIBIDORES DE LA SÍNTESIS DE ADN (1) Modificaciones de sacáridos
Aciclovir/Acicloguanosina Las VHS-1, VHS-2 y VVZ son susceptibles al aciclovir.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
 Terminación de cadena
Terminación de cadenaFigura 8 |
El aciclovir es efectivo contra las queratitis de herpes simplex, VHS latente, erupciones febriles (Herpes labiales), herpes genital. Los mutantes resistentes al aciclovir son un problema luego de uso a largo plazo y han mostrado que resultan de cambios en la timidina cinasa o en el gen de la polimerasa. Existe una prodroga del aciclovir llamada Valaciclovir ((VACV), Zelitrex®, Valtrex®) que es un éster L-valina del fármaco. Esta puede ser tomada vía oral. Penciclovir Famciclovir
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
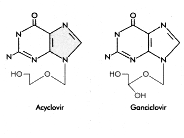 Aciclovir Ganciclovir
Aciclovir GanciclovirFigura 9 |
Ganciclovir Al igual que el aciclovir, el ganciclovir tiene su diana en la ADN polimerasa y actúa como un finalizador de cadena. En las células infectadas por herpesvirus, primero es fosforilado por la timidina cinasa vírica y luego por las cinasas celulares producir una forma trifosfato de la droga, la cual es incorporada y provoca la finalización de la cadena de ADN. Sin embargo, el CMV no codifica una timidina cinasa. En vez, el ganciclovir es fosforilado por una proteína cinasa codificada por el CMV llamada UL97 que ajusta su especificidad en las células infectadas. También se adquiere selectividad puesto que la polimerasa viral tiene una afinidad 30 veces mayor por el ganciclovir que la enzima del huésped. Normalmente, el ganciclovir se administra de modo intra-venoso a una dosis de 10mg/Kg./día u oralmente a una dosis de 3000mg/día. Usualmente se utiliza en retinitis por CMV en los pacientes con SIDA que tengan un implante intraocular (o sea, intravítreo) llamado Vitrasert. Este contiene 4.5 mg de ganciclovir para terapia local.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
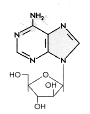 Ara-A
Ara-AFigura 10 |
Arabinósido de adenosina Nombre químico: 9-beta-D-Arabinofuranosil-9H-purin-6-amina Otros nombres: Vidarabine, Ara-A - figura 10 El Aciclovir y el Ganciclovir son finalizadores de cadena porque no tienen un anillo sacarósido completo; está faltante el grupo 3' -OH apropiado para formar un enlace fosfodiéster durante la elongación del ADN. El arabinósido de adenosina tiene un azúcar complete pero es arabinosa en vez de ribosa. El fármaco tiene muchos efectos colaterales y sólo es utilizado en enfermedades potencialmente letales. Además, es fácilmente desaminado en la circulación sanguínea a una forma menos efectiva, la ara-hipoxantina. |
||||||||||||||||||||||||||||||||||||||||||||||||||||
 AZT AZTFigura 11A |
Zidovudina Nombre químico: 3′-azido-2′,3′-dideoxitimidina Otros nombres: Azidotimidina, AZT, Retrovir® - figura 11A Este fármaco también es un finalizador de cadena. Es fosforilado por una cinasa celular y por tanto puede ser utilizado contra los virus que no posean su propia timidina cinasa (i.e. el VIH). La transcriptasa inversa (ADN polimerasa ARN-dependiente) es más sensible al fármaco que la ADN polimerasa ADN-dependiente de los humanos, dando esto cuenta de su especificidad, pero hay muchos efectos de toxicidad. Es utilizado como una droga anti-VIH tipos 1 y 2 (vea Capítulos sobre VIH). Debido a la función de la ARN polimerasa II en la síntesis del genoma de los retrovirus y la consecuente alta tasa de mutación del mismo, la presión selectiva por la presencia del fármaco rápidamente conlleva al surgimiento de mutantes víricos resistentes. Todos estos tienen mutaciones en la transcriptasa inversa. Dado el surgimiento de las formas mutantes, la AZT se administra en combinación con otros fármacos. |
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Cidofovir El fármaco se administra en forma de fosfonometoxi-nucleósido y es fosforilado dos veces intracelularmente a una forma activa, difosfato mediante dos cinasas celulares (cinasa del monofosfato del nucleósido de pirimidina y cinasa del difosfato del nucleósido de pirimidina. No hay una cinasa vírica implicada, en contraste con el aciclovir el cual es administrado en forma de nucleósido y el primer fosfato es añadido por una timidina cinasa viral). El
Cidofovir inhibe la polimerasa de ADN de un número de virus a
concentraciones que son sustancialmente más bajas que las necesitadas
para inhibir las polimerasas de ADN humanas. Es activo contra el
herpesvirus con menos efectos secundarios que el ganciclovit aunque
muestra nefrotoxicidad, entre otros efectos adversos. Debe ser
administrado junto con probenecid para bloquear la secreción tubular
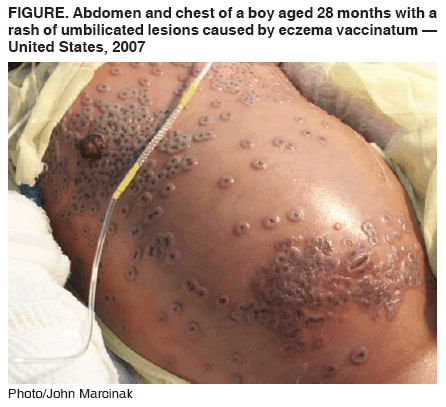
renal del mismo. El Cidofovir fue recientemente (Marzo 2007) usado (junto con una droga experimental, ST-246) en el tratamiento de un caso de eczema vaccinatum en un niño de dos años. Esta es una consecuencia secundaria inusual de la vacuna de la viruela en la cual el virus vaccinia vivo en la vacuna es transmitido a los contactos del vacunado que son usualmente inmunocomprometidos. En este caso, debido al eczema, el virus pudo penetrar la piel del paciente y replicarse, inicialmente causando una erupción dérmica difusa y luego bullas con un hoyuelo central lo cual es indicativo de infección por vaccinia. La erupción rodeaba el 50% de la piel queratinizada del paciente. Aunque el eczema vaccinatum puede ser fatal, el paciente fue dado de alta luego de 48 días en el hospital.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
FUENTES EN
LA RED (en ingles) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
CASE REPORT |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
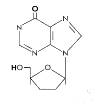 DDI
DDIFigura 12 |
Otras modificaciones del grupo sacarósido: Didesoxinosina
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
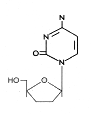 DDC
DDCFigura 13 |
Zalcitabina
Stavudine
Lamivudina
Abacavir
Emtricitabins
Tenofovir disoproxil fumarato
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
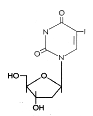 IDU
IDUFigura 14 |
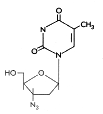
(2) Modificaciones de Base Estos son análogos de la pirimidina que son incorporados al ADN mediante la polimerasa de ADN vírica. Forman pares de bases inestables y mal-traducciones resultando en proteínas mutantes. Son inhibidores competitivos de la polimerasa viral de ADN luego de la fosforilación intracelular. Bromovinil desoxiuridina (Brivudin) Nombre químico: (E)-5-(2-bromovinil)-2′-desoxiuridina, bromovinildeoxiuridina Otros nombres: BVDU, Zostex®, Zonavir®, Zerpex®. El BVDU es usado para el tratamiento del VHS (tipo 1) y el VVZ. El fármaco es inicialmente fosforilado por la timidita cinasa viral, de ahí su especificidad. Es usado en diferentes infecciones por VSV y VVZ incluyendo la queratitis por VSH y el herpes genital. Puede ser administrado por vía oral o tópica. Yodo-desoxiuridina (Idoxuridina) Nombre químico: 5-iodo-2′-desoxiuridina Otros nombres: IDU, IUdR, Herpid®, Stoxil®, Idoxene®, Virudox® - figura 14 Este es similar al BVDU y es actualmente usado mayormente en gotas oftálmicas o en cremas tópicas para la queratitis por VSH. Trifluorotimidina (Trifluridina) Nombre químico: 5-trifluorometil-2′-deoxiuridina Otros nombres: TFT, Viroptic®. - figura 15 Este es similar en su mecanismo de acción al BVDU y al IDU. También se activa mediante la timidita cinasa viral. El TFT es usad como crema tópica o en gotas oftálmicas para queratitis por VHS. |
||||||||||||||||||||||||||||||||||||||||||||||||||||
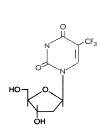 Trifluorotimidina
TrifluorotimidinaFigure 15 |
(3) Inhibidores no-nucleósidos de la transcriptasa inversa (figura 16) Debido a los inconvenientes con el AZT y los otros análogos de nucleósidos en el tratamiento del VIH, el interés en torno a otro abordaje ha ido creciendo, buscándose inhibir la misma enzima, transcriptasa inversa. Fármacos alternativos podrían ser de utilidad en terapias combinadas puesto que hay un número limitado de mutaciones que la transcriptasa inversa puede sufrir sin perder su función. Claramente, las mutaciones que dan resistencia a los inhibidores no-nucleósidos no competitivos de la transcriptasa inversa estarían en una localización diferente de la mutación que provoca que la enzima sea resistente a los análogos nucleósidos competitivos. Los inhibidores no-nucleósidos son los inhibidores selectivos de la transcriptasa inversa más potentes que se tienen, trabajando a concentraciones nanomoleculares. Tienen una toxicidad mínima en evaluaciones con cultivos celulares (actividad anti-viral a una concentración 10,000 a 100,000 veces menor que la citotóxica) y han demostrado que actúan sinergísticamente con los análogos de nucleósidos como el AZT. Más aun, actúan contra el VIH resistente a los análogos de nucleósidos. Por tanto, estos fármacos tienen un índice terapéutico alto y también presentan buena biodisponibilidad de modo que las concentraciones anti-virales son rápidamente logradas. Son inhibidores no competitivos de la transcriptasa inversa que como diana tienen lugares alostéricos en la molécula de transcriptasa inversa. |
||||||||||||||||||||||||||||||||||||||||||||||||||||
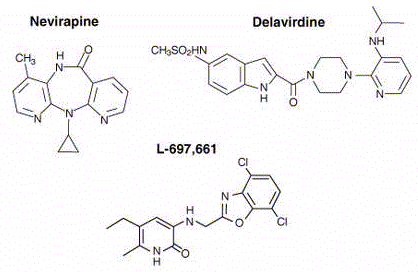 Inhibidores no competitivos de la transcriptasa inversa
Inhibidores no competitivos de la transcriptasa inversaFigura 16 |
Actualmente hay toda una colección de estos agentes que son químicamente distintos:
Nevirapina
Delavirdina |
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
 Efavirenz
(Sustiva)
Efavirenz
(Sustiva)Figura 17 |
Efavirenz
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Foscarnet Figura 18 |
4) Otros inhibidores no nucleósidos de la polimerasa
Foscarnet
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCUTRA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
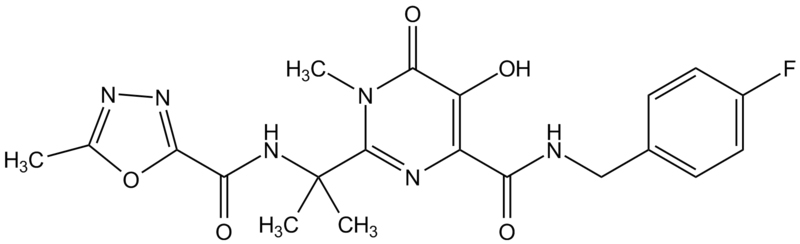 Isentress (Raltegravir)
Isentress (Raltegravir)Figura 19a
|
INTEGRACIÓN DEL ADN Los retrovirus copian su genoma de ARN a ADN usando la transcriptasa inversa. El ADN puede permanecer como un provirus circular o puede ser integrado al ADN celular. Este ultimo es necesario para la transcripción a ARN genómico y mensajero. Por tanto, la integración es necesaria para la replicación viral La integración del ADN viral es efectuada por la enzima integrasa que es codificada por el gen pol. La necesidad de integración para la replicación significa que la integrasa sería una diana selectiva de los fármacos. Recientemente, se aprobó un inhibidor selectivo de la integrasa. Raltegravir
Otros nombres:
Isentress®,
MK-0518 - figura 19a INHIBIDORES DE LA SÍNTESIS DE ARN
Ribavirina Una presentación en aerosol es usada contra el VSR (virus sincitial respiratorio) y el fármaco se usa de modo endovenoso contra la fiebre de Lassa. N.B. Ribavirina puede antagonizar los efectos del AZT como fue encontrado en algunos estudios iniciales de combinaciones terapéuticas contra el VIH.
Neplanocin
A
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
ENZIMAS DE CLIVAJE DE ARN Las ribozimas son moléculas de ARN que tienen propiedades catalíticas dentro de las cuales está el clivaje específico de ácidos nucleicos. La heptazima es una ribozima que rompe el ARN de la hepatitis C en regiones altamente preservadas (así reduciendo la posibilidad de desarrollo de resistencia). Reconoce y rompe todos los tipos conocidos del virus de la hepatitis C, finalizando así la replicación viral. La heptazima no ha tenido éxito en ensayos clínicos.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
INHIBIDORES DE LA SÍNTESIS PROTÉICA Se ha hecho poco progreso en el desarrollo de fármacos que inhiban la síntesis viral de proteínas puesto que los virus usan los mecanismos de traducción de la célula huésped. No obstante, un fármaco de esta clase está disponible. Fomivirsen
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
 El proceso de actividad de la proteasa de retrovirus en el que la
proteasa inicia como parte de la poliproteína POL y luego cliva la
poliproteína
El proceso de actividad de la proteasa de retrovirus en el que la
proteasa inicia como parte de la poliproteína POL y luego cliva la
poliproteínaFigura 20 |
INHIBIDORES DEL PROCESAMIENTO DE PROTEÍNAS Inhibidores de la proteasa Muchos virus han de clivar (romper) las proteínas que ellos producen. En el caso de las glicoproteínas de superficie, esto usualmente se lleva a cabo por una proteasa del huésped en la vía secretoria (i.e. cuerpo de Golgi). En el caso de proteínas internas, como la polimerasa o los antígenos grupo-específicos (AGGs) de los retrovirus y de algunos otros virus, hay una proteasa viral que es codificada por el gen POL (figura 20). Los inhibidores dirigidos a sitios activos del la aspartilproteasa del VIH han sido desarrollados puesto que esta enzima no es similar a las enzimas proteolíticas conocidas del huésped y por tanto estos inhibidores deberían mostrar especificidad para proteínas virales. La acción de la proteasa del VIH es crucial para la infectividad viral. Ahora se tiene la promesa de un régimen terapéutico que pueda suprimir indefinidamente el desarrollo de la enfermedad. (vea también el capítulo de Terapia anti-VIH) Los inhibidores anti proteasa de VIH son todos análogos de sustrato (figura 22). Cuando son utilizados individualmente pueden llevar a la reducción de las cargas virales de una 30a y una 100ª de veces por debajo del valor inicial pero las dosis sub-óptimas de estos inhibidores, cuando son usadas solas, pueden resultar en una pérdida de la supresión luego de varios meses y de una acumulación de múltiples mutaciones en el gen de la proteasa proveyendo así a la droga de una alto nivel de resistencia. No obstante, es de notar que los pacientes con supresión sostenida no desarrollan mutaciones resistentes. Esto parece ser debido a que la replicación ha de ser mantenida bajo la presión selectiva de la droga para el desarrollo de dichas mutaciones.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
FUENTES EN LA RED
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
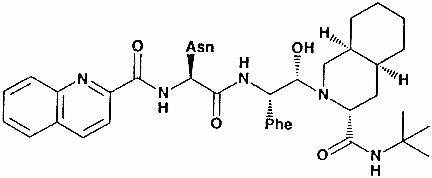 Saquinavir
Saquinavir
Figura 21 |
Saquinavir
(SQ)
Ritonavir
Este fármaco reduce los eventos delimitantes del SIDA y la mortalidad en un 58% comparada con placebo. Casa náusea en 25% de los pacientes. Es usado como parte de una terapia antiretroviral triple altamente activa (HAART).
Indinavir
El Indinavir más dos fármacos anti – transcriptasa inversa (HAART) disminuyen el VIH a un punto tal que la PCR no puede detector el virus en un 85% de los pacientes
Amprenavir
Este es otro inhibidor de la proteasa usado en la terapia HAART de combinación. Nelfinavir Lopinavir El Lopinavir es administrado en combinación con Ritonavir, otro inhibidor de la proteasa a una proporción 4/1. También, es usado como parte de la HAART. Atazanavir Bevirimato
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
Terapia antiretroviral altamente activa (HAART) Las terapias de combinación (cócteles triples de fármacos, HAART) son muy efectivos y pueden reducir la carga viral de los pacientes a niveles por debajo de los detectables sugiriendo que la replicación del VIH ha cesado. Uno de estos cócteles HAART consiste en zidovudina (AZT) , lamivudina (3TC), ambos análogos nucleósidos de la transcriptasa inversa, e Indinavir, un inhibidor de proteasa. Los niveles de ARN viral antes del tratamiento, que pueden ser tan altos como 11 millones de copias por ml, son reducidos a niveles no detectables en unas cuantas semanas por esta combinación farmacológica (se pueden medir hasta 20 copias /ml) (figura 23). La evidencia sugiere que NO hay replicación viral en estos pacientes y esto es mantenido por varios años. Cuando se detiene el tratamiento, el virus vuelve porque ha estado latente en células T de memoria y posiblemente en otras células también. Otra combinación farmacológica triple consiste en dos análogos nucleósidos de la transcriptasa inversa (tenofovir, (R)-9-(2-Fosfonilmetoxipropil)adenina) y emtricitabina (2',3'-Didesoxi-5-fluoro-3'-tiacitidina) más el inhibidor no-nucleósido de la transcriptasa inversa, efavirenz (Sustiva). El problema con todas estas complicados regímenes farmacológicos es la complianza. Los componentes de la terapia HAART deben ser tomados a diferentes horas, a veces en medio de la noche así como durante el día y deben de ser ingeridos con diferentes alimentos. Por ejemplo, si no se toma el saquinavir en las siguientes dos horas postprandiales de una comida rica en grasa, ésta no se absorberá. Por otro lado, el Indinavir debe ser ingerido prácticamente en ayunas. En los pacientes que no se toman los tres fármacos durante una semana, hay un incremento marcado de la carga viral. La no complianza con la terapia de inhibidores de la proteasa es un problema serio dado que el nuevo virus que surge es resistente a los inhibidores que se están tomando y también a otros inhibidores de la proteasa. Esto es un gran problema puesto que los nuevos mutantes resistentes pueden ser transmitidos a otros. Por tanto, si se conoce que un paciente no se compromete el/ella probablemente no debe ser introducido a estos fármacos puesto que la resistencia puede surgir tan rápidamente y puede ser transmitidas a allegados. La terapia HAART es muy costosa, por ejemplo la combinación de zidovudina/inhibidor de proteasa cuesta US$12,000 por año.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
 Complejo Proteasa del VIH-1 con un inhibidor péptidomimético
macrocíclico
Complejo Proteasa del VIH-1 con un inhibidor péptidomimético
macrocíclicoRequiere un plug-in de Chime. Adquiera Chime aquí – haga clic en la imagen para abrir el archivo Figura 22B
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Este
diagrama derivó de una cristalografía de rayos X y enseña la
aspartilproteasa dimérica del VIH (lazos). Los residuos de aspartato se
muestran como ‘pelotas y bates’. Note que cuatro aspartatos forman
cúmulos en los sitios activos de la enzima. Un inhibidor de protasa se
muestra insertado en sitio activo Figura 24 |
¿Podemos
curar la infección por VIH con terapia farmacológica?
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
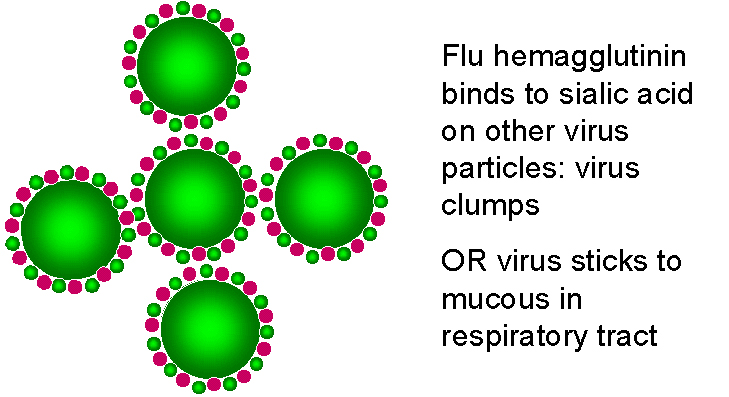
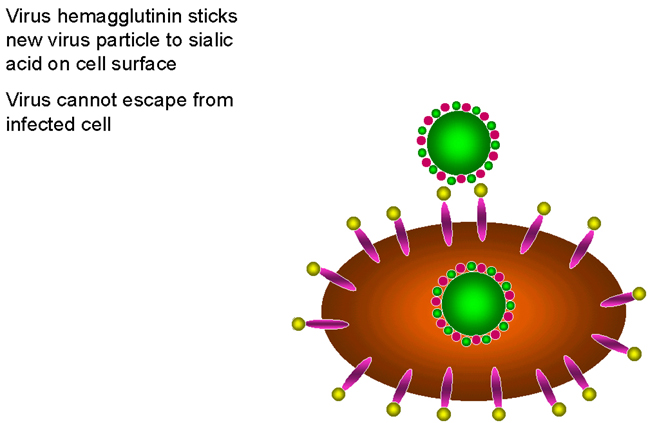 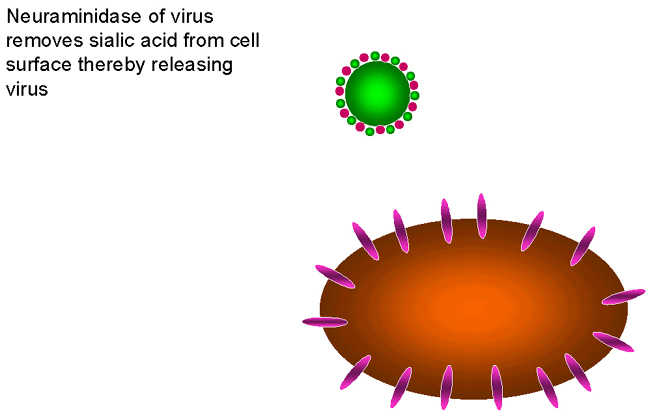 Los requerimientos de neuraminidasa en el ciclo vital del virus de la
influenza
Los requerimientos de neuraminidasa en el ciclo vital del virus de la
influenzaFigura 24
|
INHIBIDORES DE LA MODIFICACIÓN DE PROTEÍNAS
Zanamivir
Oseltamivir OTROS OBJETIVOS En el ciclo vital de los retrovirus, el enfocamiento de proteasas específicas que son necesarias para la formación de una partícula vírica infecciosa ha sido particularmente exitoso en el desarrollo de nuevos fármacos. En etapas tempranas, los inhibidores de la transcriptasa inversa también fueron exitosos pero los análogos de nucleósidos causaban severos efectos adversos dado que también inhibían la ADN-polimerasa del huésped. En contraste, los inhibidores no-nucleósidos de la transcriptasa inversa muestran excelentes índices terapéuticos. En cualquier caso, sin embargo, la monoterapia conlleva al rápido surgimiento de mutantes resistentes. Hay muchos otros objetivos en investigación para la intervención de los ciclos vitales de muchos virus, por supuesto, la meta es la especificidad. En el caso de los retrovirus, además de aquellos fármacos descritos anteriormente, los inhibidores de la integrasa están siendo extensivamente estudiados pero ninguno ha logrado todavía ser establecido en tratamientos clínicos de rutina. Otros abordajes interesantes pueden ser encontrados en las páginas siguientes |
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
FUENTES EN
LA RED (en inglés) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ESTRUCTURA MOLECULAR |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Derechos de autor 2007 The Board of Trustees of the University of South Carolina
|
||||||||||||||||||||||||||||||||||||||||||||||||||||

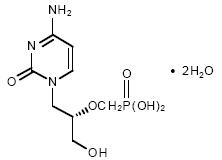
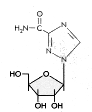 Ribavirin
Ribavirin