![]()

Dr. Margaret Hunt
VIROLOGÍA – CAPÍTULO VENTITRÉS
ENFERMEDADES VIRALES LENTAS DEL SISTEMA NERVIOSO
Traducido por :
Sarah M. Castillo - Jorge, Clinica Corominas
Santiago, Rep. Dominicana
VIDEOCONFERENCIA
EN INGLÉS
Revisado Octubre 2006
Editado e ilustrado por Dr. Richard Hunt
LECTURA: Murray et al., Microbiología, 5a. Ed., Capítulo 67 (priones)
OBJETIVOS
Introducción a las enfermedades subagudas/crónicas del sistema nervioso central.
Propiedades de los agentes no convencionales asociados con enfermedades subagudas del sistema nervioso central
GENERAL
El término "Infecciones de Virus Lentos" se refiere al tiempo de la ENFERMEDAD, no a la tasa de crecimiento del virus. Estas enfermedades tienen un periodo de incubación prolongado (el cual puede ser de meses o años), y curso clínico prolongado, extenso.
Las enfermedades de virus lentos pueden ser causadas por virus convencionales o por virus no convencionales (también llamados agentes no convencionales o agentes/virus atípicos).
Los síntomas asociados con enfermedad de virus lenta/por priones del sistema nervioso tienden a tener múltiples manifestaciones neurológicas. Los diferentes pacientes pueden tener síntomas muy diversos.

Figura 1
Cerebro en LMP
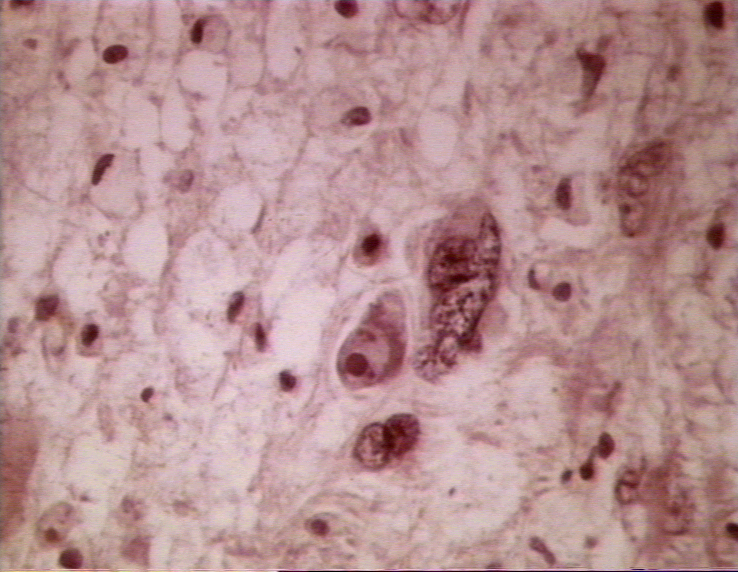 Figura 2
Figura 2
Cerebro con LMP © Archivo de Imágenes Biomédicas de Bristol. Usado con
autorización
VIRUS CONVENCIONALES
Leucoencefalopatía multifocal progresiva (LMP)
Esta es una enfermedad rara, progresiva y fatal del SNC que elimina a los oligodendrocitos (figuras 1 y 2). Resulta en la pérdida de memoria, pérdida de coordinación, problemas mentales, problemas visuales, etc.
La enfermedad es causada por ciertos miembros de la familia de los poliomavirus, frecuentemente por el virus JC. La serología indica que la exposición al virus JC es muy común, pero que la LMP es rara. Los pacientes que la desarrollan regularmente tienen alguna anomalía del sistema inmune. La LMP se desarrolla en casi 5% de los pacientes son SIDA. En el SIDA, el tratamiento HAART puede ser capaz de estabilizar a, por lo menos, una parte de estos pacientes y su cuadro neuroradiológico puede mejorar. No obstante, no todos los pacientes VIH-positivos con LMP manifiestan una respuesta obvia a la terapia HAART en lo que respecta a la LMP. La LMP puede deberse a una reactivación del virus JC latente, probablemente en los riñones. También hay mucho virus en el cerebro.
Otro polioma virus, el virus BK, puede establecer una infección latente en los riñones y ser reactivado bajo condiciones de inmuno supresión pudiendo causar infecciones severas del tracto urinario. No se asocial con LMP. Se ha sugerido recientemente (2006) que el virus BK juega un rol en el cáncer de próstata.
Pan encefalitis esclerosante subaguda (PEES)
Esta enfermedad es una rara complicación del virus del sarampión y se desarrolla de 1 a 10 años después de la infección inicial. Es progresiva y fatal y se caracteriza por deterioro mental y motor. Los factores de riesgo incluyen el haber adquirido sarampión a una edad temprana.
La PEES se asocia con formas defectuosas del virus en el cerebro y por tanto es difícil aislar el virus infeccioso de los pacientes. La incidencia ha disminuido desde la introducción de la vacuna anti-sarampión.
Pan encefalitis progresiva de la rubella (PPR)
PPR es una consecuencia muy rara de una infección por el virus de la rubella y también resulta en deterioro mental y motor. La infección inicial es con frecuencia congénita o temprano luego del nacimiento y la instauración del la PPR se da a los 8 a 19 años de edad. El curso de la enfermedad se puede extender por varios años.
Otras infecciones de virus lentos
El Virus de la Inmunodeficiencia Humana y el SIDA. Refiérase a la sección de VIH/SIDA
Rabia. Refiérase a la sección de rabia
PRIONES
 Figura 3
Figura 3Comparación de las propiedades de los virus convencionales y los priones
Algunas enfermedades lentas del sistema nervioso central son causadas por un grupo de agentes inusuales, cuya naturaleza es aún controversial. En ciertas cosas los agentes se parecen a los virus convencionales: Son muy pequeños, son agentes filtrables que requieren de un huésped para crecer. No tienen capacidad para generar energía o para la síntesis proteica.
En otras cosas, sin embargo, son un tanto diferentes a los virus; por ejemplo, no hemos visto evidencia de partículas víricas en tejidos infectados o preparaciones purificadas de material infectado. Nadie ha podido probar que estos agentes contienen ácido nucleico. Si contienen acido nucleico, es probable que sea demasiado pequeño y con muy poca capacidad codificadora. Estos agentes tienen una resistencia inusual a los tratamientos comúnmente utilizados para inactivar los virus. (Figura 3 y 4).
 Figura 4
Figura 4
Propiedades de los virus no convencionales
 Figura 5
Figura 5
Encefalopatías espongiformes transmisibles
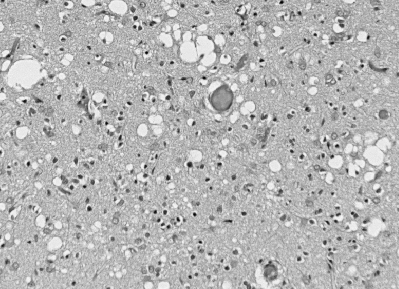 Figura 6
Figura 6
Cambio espongiforme en la enfermedad de Creutzfeldt-Jakob consiste en
vacuolas redondas dentro del neuropilo lo que ocurre tanto en solitario
como en grupos confluentes, distorsionando la citoarquitectura
cortical. © Unidad de Vigilancia de la Enfermedad de Creutzfeldt-Jakob
en el Reino Unido
Estos virus o agentes no convencionales con frecuencia se llaman ‘priones’ – puesto que hay proteína presente en las preparaciones purificadas de material infectado y el tratamiento que destruye la proteína destruye su infectividad. En contraste, los tratamientos que destruyen ácido nucleico no destruyen su infectividad. Esta proteína es conocida como PrP (proteína de prion). La pregunta de si el ácido nucleico es parte de estos agentes todavía es controversial; muchas personas creen que el material infeccioso es solo la proteína.
Estos agentes:
· Causan enfermedades confinadas al SNC
· Tienen un periodo de incubación prolongado
· Muestran un curso clínico lento, progresivo, fatal
· Manifiestan una encefalopatía espongiforme
· Característicamente resulta en vacuolación de las neuronas
· Pueden causar la formación de agregados fibrilares, que contienen PrP y tienen características amiloideas
Las enfermedades causadas por estos agentes (encefalopatías espongiformes transmisibles (figura 5)) son relativamente raras en humanos, pero se especula que puede ser más común de lo que se creía anteriormente y que pueden estar implicadas en lose estudios de otras enfermedades degenerativas del SNC. Pueden ser adquiridas, heredadas, u ocurrir esporádicamente.
Scrapie (Tembladera)
El Scrapie es una enfermedad de las ovejas. Resulta en cambios de comportamiento, progresa a tremores, ataxia (falla de coordinación muscular), debilitación y muerte. Es una enfermedad transmisible.
Kuru
Kuru (o enfermedad de la risa) es una enfermedad de los humanos. Causa tremores y ataxia y, con frecuencia, en estadios tardíos, demencia. Se transmite por ritos de muerte incluyendo autopsias y canibalismo en la población de la tribu Fore en Papua/Nueva Guinea. Desde que cesaron estas prácticas nadie que haya nacido ha adquirido el Kuru. No hay evidencia de transmisión a fetos, de transmisión vía lactancia o contacto social íntimo.
FUENTES EN LA RED
NINDS Página informativa del
Kuru
Enfermedad de Creutzfeldt-Jakob
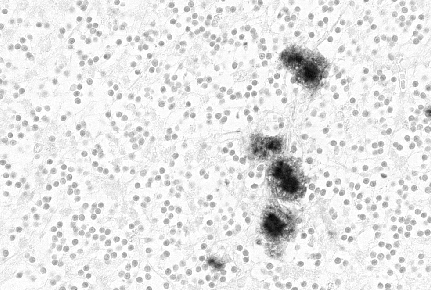 Figura 7
Figura 7Inmunotinción de la proteína de un prion mostrado placas amiloideas. © Unidad de Vigilancia de la ECJ en el Reino Unido
Enfermedad de Creutzfeldt-Jakob (ECJ)
La ECJ es una enfermedad del hombre que resulta en demencia y también tremores y pérdida de la coordinación motora. Se dan de 1 a 3 casos por millón de habitantes por año pero los casos pueden no ser diagnosticados. La enfermedad puede transmitirse animales en el laboratorio. Aunque se ha visto que la enfermedad se desarrolla en las edades de los 16 a los 80+ años, es usualmente observada en personas de 50 a 70 años de edad. 10% de los casos son familiares lo que sugiere que hay algún gen que hace al individuo susceptible de la ECJ.
El medio usual de transmisión no se conoce pero muchos casos son esporádicos y no hay evidencia de transmisión de persona a persona. La ECJ puede ser transmitida por manipulación médica: transplantes de cornea, transplantes de dura madre, uso equipo mal esterilizados en neurocirugía (ya los procedimientos de esterilización han sido cambiados para prevenir esto), administración de hormona del crecimiento de cadáveres (ahora se usa un vector de ADN recombinante para sintetizar la hormona de crecimiento).
FUENTES EN LA RED
EBE
CDC
Página Oficial de la Enfermedad de La Vaca Loca
Noticias
del BSE
Encefalopatía bovina espongiforme y una variante de la enfermedad de
Creutzfeldt-Jakob: Antecedentes, Evolución y Asuntos Actuales
EBE Tutoría del Dr. Alan Cann, Universidad de Leicester
¿Cuántas personas han contraído vCJD (tablas del Reino Unido)
Nueva variante de la ECJ (EBE humana); nvCJD, vCJD
Una forma de ECJ ha sido reportada, predominantemente en los Reino Unido, en pacientes más jóvenes (frecuentemente de menos de 40; con una edad promedio a su muerte de 28 años) que los de ECJ (edad promedio a la muerte: 68 años) (figura 8A). Esta enfermedad es difiere también de la ECJ en que los pacientes tienden a presentar problemas siquiátricas y el curso de la enfermedad tiende a ser más extenso. Quienes la padecen pueden eventualmente mostrar uno o todos los síntomas descritos anteriormente para las otras enfermedades por priones. La patología fue primero vista en 1996 y hay evidencia muy sugestiva de que se asocial con la exposición a carne de res contaminada con EBE. Ya han sido implementadas fuertes medidas de control de EBE. Las autopsias revelan una apariencia neuropatológica distintiva y más depósitos de placas de tipo amiloidea y de PrP (proteína de prion) (figura 7 y 9).
| Diferencias entre la ECJ clásica y su variante | Número de casos de EBE |
Para Agosto del 2006, 162 personas habían sido reportadas con la vCJD en el RU (figura 8b), 20 en Francia, 4 en Irlanda, 2 en los Estados Unidos y 1 en Canadá, Japón, Portugal, España y Holanda (las personas de Canadá, Japón y EU probablemente fueron expuestas durante su estancia en el RU).
Aún no sabemos si estamos presenciando el inicio de una gran epidemia o si esta es la mayor cantidad de casos a ser vistos de esta patología. También hay preocupaciones porque con la vCJD parece haber más infección de tejido periférico, especialmente linforeticular, que con la ECJ clásica. Esto provoca la pregunta sobre la esterilización de los instrumentos quirúrgicos etc. y la posibilidad de contagio iatrogénico. Esto es también una de las razones por las que en los Estados Unidos son cautelosos y protectores de los suplementos sanguíneos (mediante el rastreo de aquellos que han pasado un tiempo considerable en el RU o en Europa). Existe la probabilidad de que los agentes de la vCJD se transmitan por transfusiones sanguíneas en dos casos en el Reino Unido. En ambos casos, la sangre no había sido depletada de leucocitos, y se cree que la depleción de leucocitos puede disminuir las probabilidades de transmisión por transfusión sanguínea.
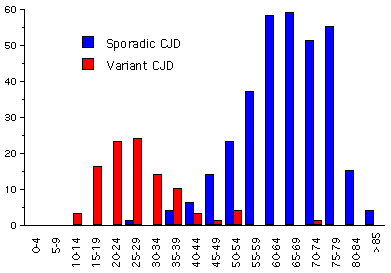 Figura 8A
Figura 8ADistribución etárea de la ECJ en RU: ECJ y vCJD 1994-2001 USDA
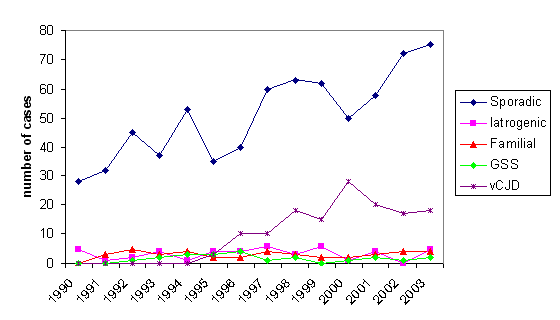 Figura 8B
Figura 8BNúmero de muertes por ECJ en el RU 1990-2003
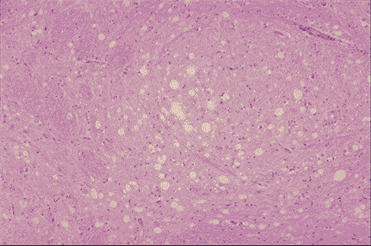
 Figura 9A
Figura 9A
Medula de res afectada con EBE: se ven vacuolas en una neurona y en los
neuropilos. Proliferan astrocitos con núcleos pequeños. No hay infiltrado de
células inflamatorias en el cerebro. Izquierda x100, Derecha x200 Dr. M.
KUBO NIAH, Japón
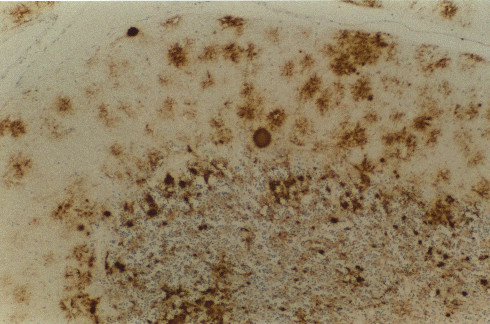 Figura 9B
Figura 9B
Inmunocitoquímica para la PrP en el cerebelo muestra placas tipo-kuru de
fuerte tinción (centro) con placas más pequeñas en la capa granular y
abundante deposición pericelular en la capa molecular © Unidad de
Vigilancia de la ECJ en el RU /The Lancet
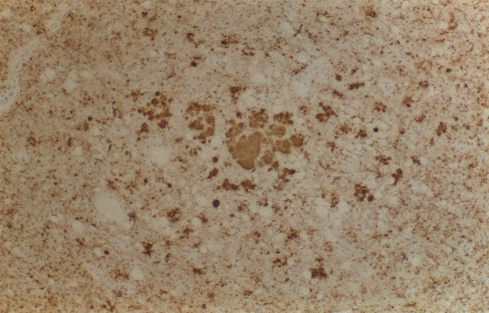 Figura 9C
Figura 9C
Inmunocitoquímica de la PrP en el tálamo muestra varias placas grandes
multicéntricas (centro) con deposición perivacuolar y sináptica en el
neuropilo circundante © Unidad de Vigilancia de la ECJ en el RU /The
Lancet
Síndrome de Gerstmann-Sträussler-Scheinker
El síndrome de Gerstmann-Sträussler-Scheinke (GSS) es una enfermedad del hombre y tiene síntomas similares al Kuru. Es una enfermedad familiar con frecuencia considerada como un subtipo de ECJ de transmisión genética. Esta enfermedad es transmisible a animales de laboratorio.
Insomnio Familiar Fatal
El Insomnio Familiar Fatal es una enfermedad de humanos y resulta en un insomnio progresivo incurable, pérdida de los ritmos circadianos, desórdenes endocrinos, desórdenes motores, demencia. También es familiar (heredada) y puede transmitirse a animales de laboratorio. En esta forma de la enfermedad, parece que el hipocampo actúa como un órgano diana inicial.
PROTEÍNA DE PRION
Las preparaciones de material infectado altamente purificado contienen grandes cantidades de PrP (figura 9 y 10). Esta es codificada por un gen celular del huésped y es una proteína de superficie anclada a un glicofosfolipidilinositol (GPI). Su función normal no se conoce. La forma infecciosa tiene la misma secuencia de amino ácidos y las mismas modificaciones post traducción que la forma normal, pero tiene una conformación diferente en el tejido enfermo. La forma normal contiene muchos alfa-hélices, mientras que la forma asociada a patología tiene muchas hojas beta-plegadas. La forma asociada a enfermedad se conoce como PrPRES puesto que es más resistente a proteasas o como PrPSC puesto que primero fue aislada de infecciones de scrapie.
¿Porqué una proteína es infecciosa?
Una hipótesis es que la forma resistente puede convertir la forma normal en resistente, que entonces será capaz de convertir otras formas normales de la proteína en formas resistentes; así la tasa de conversión se amplifica gradualmente a medida que la concentración de las formas resistentes aumenta. Pare de esta conversión parece darse extracelularmente.
Casos adquiridos
Los casos adquiridos pueden deberse a infecciones con la forma resistente, lo cual entonces convierte las formas normales de la persona en PrPSC, y así el proceso gradualmente se amplifica como se mencionó antes.
Casos esporádicos
Los casos esporádicos pueden deberse a conversiones espontáneas de la forma normal a la forma resistente, y este proceso se amplifica a medida que la forma resistente convierte más formas normales en resistentes. Los casos esporádicos también pueden deberse a mutaciones somáticas, que hacen que la PrP sufra una conversión espontánea a la forma resistente, o puede ser adquirida por un mecanismo desconocido.
Casos familiares (hereditarios)
En los casos familiares, se han visto mutaciones del gen de la PrP. En la forma hereditaria d esta enfermedad, la forma mutada de la proteína puede tener más probabilidad de cambio espontáneo a la forma resistente y luego el mismo proceso de reclutamiento ocurre. Por lo menos para algunas de estas mutaciones, parece que todo aquel que hereda un gen mutante eventualmente desarrolla ECJ/GSS si viven lo suficiente. La naturaleza de estas mutaciones en la forma heredada puede afectar el curso clínico de la enfermedad.
RESPUESTA INMUNE
Estos virus/agentes no convencionales no causan una respuesta inflamatoria. No inducen el interferón. No hay una respuesta de anticuerpos contra estos agentes. Por tanto no hay posibilidad de rastreo de la exposición a estos agentes por detección de anticuerpos.
TRATAMIENTO
Hasta la fecha, las enfermedades por priones han sido invariablemente fatales. La ECJ clásica generalmente resulta en la muerte algunos meses después de que los síntomas se hacen obvios. El tiempo promedio desde que los síntomas se hacen obvios hasta la muerte en la VCJD es más largo – como de dieciséis meses. Debido al pobre pronóstico de los pacientes con ECJ, varias drogas han sido evaluadas para su eficacia, pero hasta ahora, parecen tener muy pocos efectos positivos, que si fueran reales fueran muy transitorios; más aún los efectos secundarios de estas drogas pueden ser muy serios.
Otro abordaje ha sido hecho para crear anticuerpos que inhiban la formación de priones en ratones. La parte prometedora de esto es que parece que si uno detiene la formación de más PrPSC, las células pueden desechar la PrPsc que ya se ha formado. Este abordaje todavía no ha sido intentado en humanos.
Un aspecto a recordar es que si hubiese drogas que retardaran la progresión, aún cuando no curaran la enfermedad sintomática, pueden ser de utilidad en las formas familiares, casos en los cuales podrían administrarse antes del desarrollo de los síntomas. Esta es un área muy nueva de las enfermedades por priones.
DIAGNÓSTICO
Durante el tiempo de vida, un probable diagnóstico se basa en el cuadro clínico. El EEG puede aportar evidencia respaldante en algunos casos. El amplio rango de los síntomas de del curso de la enfermedad hacen que el diagnóstico sea difícil y las enfermedades por priones con frecuencia no son diagnosticadas. El diagnóstico final usualmente se hace por evaluaciones post-mortem del cerebro. Puede hacerse biopsia cerebral. La serología es inútil puesto que estos pacientes no desarrollan una respuesta inmunológica.
En los casos de vCJD, La RMN puede ser útil en el diagnóstico. Un diagnóstico positivo puede a veces hacerse por la presencia de PrPSC en tejido linfoide periférico; por ejemplo, las biopsias tonsilares. Es posible sintetizar anticuerpos a la PrPSC usando ratas que de las que haya sido eliminado el gen de la PrP y que por tanto no toleren la proteína. Estos anticuerpos pueden ser usados en análisis de tipo Western blot si puede extraerse suficiente material de prion del paciente.
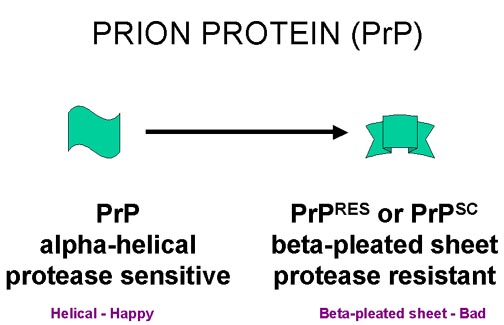
La proteína del prion PrP (codificada por
el gen celular y sintetizada en células normales) puede existir en dos
formas. En el tejido enfermo la forma resistente a proteasa (PrPsc) con
muchos pliegues beta se acumula como ‘placas amiloideas'
Figura 10
La PrPsc, la forma molecular resistente a proteasa, actúa como ‘plantilla’. Se asocia con la forma helicoidal permitiendo a esta última ser convertida a la forma resistente de pliegues beta (presuntamente mediante la disminución de barreras energéticas que normalmente previenen que esto suceda). Ahora hay dos moléculas de la forma resistente que pueden actuar como plantilla y así el proceso se acelera.
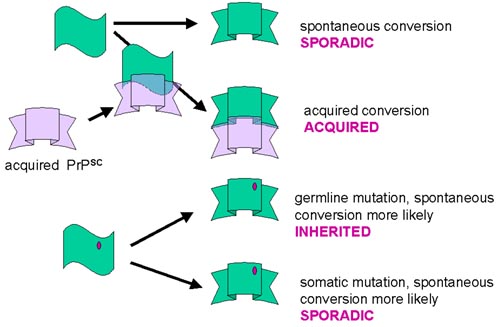 ¿Cómo puede este modelo explicar las formas esporádicas, adquiridas o heredadas
de la enfermedad? La conversión de la forma alfa helicoidal a la forma beta
plegada puede darse espontáneamente, aunque muy raro (esporádica). La
conversión puede ser catalizada por la PrPsc que proviene de alguna
fuente exógena (adquirida). Las mutaciones de la línea celular germinal
pueden crear conversiones espontáneas con más probabilidad (heredada). Las
mutaciones somáticas pueden crear conversiones espontáneas con más probabilidad
(esporádica). En este caso, la forma mutante puede iniciar el proceso de
conversión y resultar en moléculas de PrPsc que luego convertirán la
forma normal de las células adyacentes.
¿Cómo puede este modelo explicar las formas esporádicas, adquiridas o heredadas
de la enfermedad? La conversión de la forma alfa helicoidal a la forma beta
plegada puede darse espontáneamente, aunque muy raro (esporádica). La
conversión puede ser catalizada por la PrPsc que proviene de alguna
fuente exógena (adquirida). Las mutaciones de la línea celular germinal
pueden crear conversiones espontáneas con más probabilidad (heredada). Las
mutaciones somáticas pueden crear conversiones espontáneas con más probabilidad
(esporádica). En este caso, la forma mutante puede iniciar el proceso de
conversión y resultar en moléculas de PrPsc que luego convertirán la
forma normal de las células adyacentes.
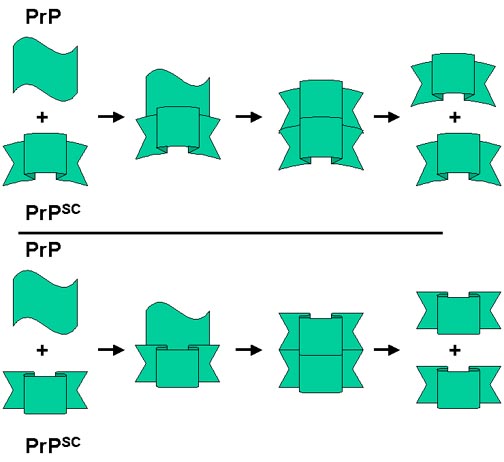 ¿Por qué hay diferencias entre las enfermedades por priones? Puede haber
diferencias sutiles entre las formas resistentes a proteasa (PrPsc)
de la proteína de prion según la fuente de la PrPsc o la
mutación implicada. Como se indica en la figura, las formas relacionadas, aunque
sutilmente diferentes, de la PrPsc convierten la forma
normal a su propia conformación. Entonces, la PrPsc final
que se acumula depende de la forma que inició el proceso.
¿Por qué hay diferencias entre las enfermedades por priones? Puede haber
diferencias sutiles entre las formas resistentes a proteasa (PrPsc)
de la proteína de prion según la fuente de la PrPsc o la
mutación implicada. Como se indica en la figura, las formas relacionadas, aunque
sutilmente diferentes, de la PrPsc convierten la forma
normal a su propia conformación. Entonces, la PrPsc final
que se acumula depende de la forma que inició el proceso.
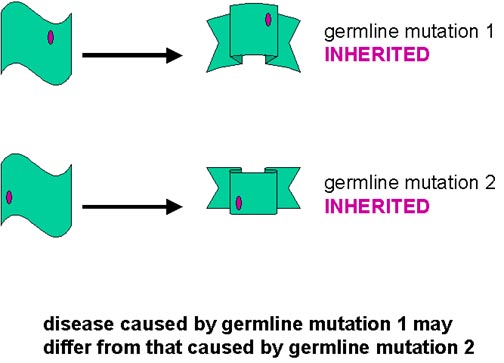 Esta explicación también puede aplicarse a las formas heredadas. Las diferentes
mutaciones predisponen a la PrPsc a adoptar
espontáneamente formas ligeramente diferentes de resistencia a la proteasa
Esta explicación también puede aplicarse a las formas heredadas. Las diferentes
mutaciones predisponen a la PrPsc a adoptar
espontáneamente formas ligeramente diferentes de resistencia a la proteasa
ENCEFALOPATÍAS TRANSMISIBLES Y OTRAS ENFERMEDADES
Las placas amiloideas se ven en otras enfermedades del SNC – pero el mayor componente de las placas amiloideas visto, por ejemplo, en la enfermedad de Alzheimer NO es el mismo que el que se ven en Kuru, ECJ, GSS. Amiloidea se refiere a sus propiedades de tinción, y muchos agregados de proteínas glicosiladas pueden tener propiedades similares de tinción.
Es posible que la forma que en al enfermedad interfiere con la función del SNC pueda señalar procesos cruciales en el SNC cuya alteración conlleva a degeneración progresiva del tejido nervioso. El entendimiento de la naturaleza de la patogénesis por priones puede ayudar a entender otras enfermedades del SNC.
Regreso a la sección de Virologia Microbiología e Inmunología on line
Derechos de autor 2008 The Board of Trustees of the University of South Carolina
Esta página se modificó recientemente en Wednesday, March 26, 2008
Mantenimiento de la pagina por Richard Hunt
Favor de reportar problemas a rhunt@med.sc.edu