|
x |
x |
|
 |
 |
|
INFECTIOUS
DISEASE |
BAKTERIYOLOJİ |
İMMÜNOLOJİ |
MYCOLOGY |
PARASITOLOGY |
VIROLOGY |
|
ENGLISH |
İMMÜNOLOJİ – BÖLÜM ONDOKUZ
İMMÜN YETMEZLİK
Abdul Ghaffar, Ph.D.
Emertius Professor of Pathology, Microbiology and Immunology
University of South Carolina
Çeviri:
Doç. Dr. Erkan Yula
İzmir, Katip Çelebi Üniversitesi, Tıp Fakültesi, Tıbbi Mikrobiyoloji
Anabilim Dalı
|
|
FRANCAIS |
|
TURKISH |
Let us know what you think
FEEDBACK |
|
SEARCH |
| |
|
|
 |
|
Logo image © Jeffrey
Nelson, Rush University, Chicago, Illinois and
The MicrobeLibrary |
|
|
|
ÖĞRENİM HEDEFLERİ
Primer ve sekonder immünyetmezlikleri bilmek
AIDS ve diğer durumlarda immün yetmezlikleri tanımak
Önemli primer immün yetmezlikleri ve özelliklerini bilmek
Lezyonun yeri ve bu yüzden oluşan immün yetmezlik arasındaki ilişkiyi
anlamak
Farklı immünyetmezliklerdeki tanı yöntemlerini bilmek. |
İMMÜN YETMEZLİK
İmmün yetmezlik, hastalık veya maligniteden korunmada
immün sistemin etkili olamamasıdır.
rimer immün yetmezlik, immün sistemin kalıtsal veya
gelişimsel defektlerinden kaynaklanmaktadır. Bu defektler doğumda
bulunmakadır ama hayatın ilerleyen dönemlerinde ortaya çıkabilir.
ekonder veya kazanılmış immün yetmezlik ise hastalık
etkenlerine maruziyet, çevresel faktörler, immun baskılanma veya
yaşlanmanın bir sonucu olarak immün fonksiyonun kaybıdır.
|
|
|
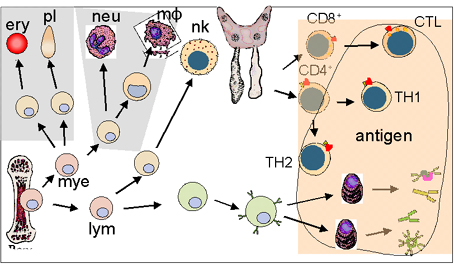
PRİMER İMMÜN
YETMEZLİKLER
Primer immün yetmezlikler immün sistemin kalıtsal
defektleridir (Şekil 1). Bu defektler spesifik veya non-spesifik immün
mekanizmalar ile olabilir. Bu yetmezlikler immün sistem gelişimi ve
farklılaşmasında sorunun bulunduğu yere göre sınıflandırılırlar. İmmün
yetmezlikli bireyler çok çeşitli infeksiyonlara duyarlıdır ve infeksiyonun
tipi immün yetmezliğin tabiatına bağlıdır (Tablo 1).
|
Tablo 1. Prime immün yetmezliğin
karakteristik enfeksiyonları |
|
Sorun |
Primary pathogen |
Primer bölge |
Klinik örnek |
|
T-hücreleri |
Intrasellüler, bakteri, virusler, protozoa,
mantarlar |
Non-spesifik |
SCID, DiGeorge |
|
B- hücreleri |
Pneumococcus,
Streptococcus, Haemophilus
|
Akciğer, deri, merkezi sinir sistemi
|
IgG, IgM yetmezliği
|
|
Enterik bakteri ve virüsler |
Gastrointestinal, nazal, göz |
IgA yetmezliği |
|
Fagositler |
Staphylococcus,
Klebsiella Pseudomonas |
Akciğer, deri, bölgesel lenf nodu |
Kronik granulomatoz hastalık |
|
Kompleman |
Neisseria, Haemophilus,
Pneumococcus, Streptococcus |
Merkezi sinir sistemi, akciğer, deri
|
C3, Factör I ve H, geç kompleman komponentleri |
|
|
|
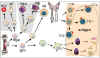 Şekil 1
Şekil 1
Primer immün yetmezliklerde gelişimsel defektler
|
| |
ÖZGÜL İMMÜN SİSTEM
Kök hücre farklılaşmasındaki defektlerin sonucu olarak
çeşitli immun yetmezlikler vardır ve bunlar T hücreleri, B hücreleri ve/veya
farklı immunglobulin sınıfları ve alt sınıflarını kapsayabilirler (Tablo
2). Kök hücrelerini içeren erken hematopoezdeki bir defekt, yaygın immün
defekte ve takibinde infeksiyonlara duyarlılığa yol açan retiküler
disgenezis ile sonuçlanır.
Bu durum genelde ölümcül olmasına rağmen çok nadirdir.
Kemik iliği transplantasyonu ile başarılı bir şekilde tedavi edilebilir.
Lenfoid seri immün defektleri
Lenfoid öncü hücreler defektli ise, T ve B hücre serilerinin her
ikisi de etkilenir ve ciddi kombine immün yetmezlik (SCID) ile
sonuçlanır. Yenidoğanlar özellikle fırsatçı mikroorganizmaların
yaptığı rekürren infeksiyonlara (bakteriyel, viral, mikotik ve
protozoal infeksiyonlar) maruz kalırlar. SCID’Lİ hastaların % 50
kadarında immün yetmezlik X’e bağımlı geçerken diğer yarısında
otozomaldır. Her ikisi de T ve B hücre immünitesinin yokluğu ve
dolaşımda T ve B lenfositlerin bulunmayışı (veya düşük düzeylerde
oluşu) ile karakterizedir. Timus gölgesi X-ray grafisinde görülmez.
X’e bağlı SCID, IL-2’nin gama zincirindeki defektten
kaynaklanır ve IL-2 aynı zamanda IL-4,-7,-11,-15 ile birlikte
lenfoid çoğalma ve farklılaşma için gereklidirler. Otozomal SCID ise
esas olarak, sırasıyla dATP veya dGTP birikimiyle neden olan ve
lenfoid kök hücrelerde toksisiteye yol açan adenozin deaminaz (ADA)
veya pürin nükleozid fosforilaz (PNP) genlerindeki defektler
nedeniyle oluşmaktadır. RAG1, RAG2 ve IL-7-alfa gibi diğer genetik
defektler de SCID’e yol açarlar. SCID’ten şüpheleniliyorsa hastaya
canlı aşı yapılmamalıdır aksi takdirde ilerleyici enfeksiyona yol
açacaktır. Tanı immünglobülin düzeylerinin ölçümü ile T ve B
lenfositleri sayımına dayalıdır. Ciddi kombine immun yetmezlik kemik
iliği transplantasyonu ile tedavi edilebilir. Son zamanlarda ADA
defektinin bulunduğu otozomal SCID’li hastalarda retroviral vektör
aracılı gen transferi ile birkaç başarılı sonuç elde edilmiştir.
SCID birçok bozukluğu
içerir
Rekombinaz aktive edici genler
T ve B hücre yetmezliğinin her ikisine de sahip hastalarda,
immünglobülin gene düzenlenmesi ve T hücre reseptörleri için
gerekli olan rekombinaz aktive edici genler(RAG1 ve 2)
bulunmaz. Bu hastalarda timus bulunmaz ve T hücre
reseptör(TCR) gen düzenlenmesinin araştırılmasıyla konur.
Anneden geçen pasif antikorlar nedeniyle B hücre defekti
bulguları erken yenidoğan döneminde gözlenmez. NK hücreler
bu hastalarda normaldir. Otozomal resesif kalıtılır.
CD3 zinciri
Bazı SCID’li hastalarda T hücreleri bulunabilir fakat TCR
ile ilişkili olan CD3 zincirinin aracı olduğu
sinyalizasyondaki defektten dolayı fonksiyonel olarak
defektiftir.
İnterlökin-2 reseptörü
IL-2 reseptörü ortak gama zinciri (IL-2Rγc) bulunmayan
hastalarda büyüme faktörleri olarak rol alan IL-2 ve diğer
sitokinlerin yaptığı sinyalizasyon önlenebilir. Bu da T, B
ve NK hücrelerin proliferasyonunda defekte yol açar.
Otozomal resesif kalıtılır
Adenozin deaminaz
Adenozin deaminaz(ADA), adenozini inozine dönüştüren bir
enzimdir. ADA enzim defekti, adenozin birikimi sonucu DNA
sentezini bozan toksik metabolitlerin oluşmasına yol açar.
Bu hastalarda T,B ve NK hücreler defektiftir. SCID’ler
otozomal resesif kalıtılır ve kök hücre nakli veya gen
tedavisi ile tedavi edilebilir.
|
Tablo 2. B ve T hücre immün yetmezlik
hastalıklarının özeti |
|
Hastalık |
T-hücre |
B-hücre
No
|
İmmünoglobülinler |
Kalıtım |
|
No. |
Fx |
IgM |
IgG |
IgA |
|
Retiküler disgenezis
|
A |
A |
A |
A |
A |
A |
u |
| CID (otozomal) |
A/L |
A/L |
A/L |
A/L |
A/L |
A/L |
a |
| SCID
(x-linked) |
A/L |
A/L |
A/L |
A/L |
A/L |
A/L |
x |
| DiGeorge's sendromu |
A/L |
A/L |
N/V |
N/V |
N/V |
N/V |
a/x |
|
Ataxia
telangiectasia |
L |
L |
L |
N/V |
L/V |
L |
a
|
|
Wiskott-Aldrich
|
?V |
L |
L/V |
L |
N |
H |
x |
| Aynı
zamanda yüksek IgE |
|
X‘e bağlı hipogamaglobulinemi |
N |
N |
L |
L |
L |
L |
x |
|
Selektif IgA eksikliği |
N |
N |
N |
N |
L/V |
L |
a/x |
|
Hiper-IgM hipogamaglobulinemi |
N |
N |
N |
H |
L |
L |
x |
|
Geçici hipogama-globulinemi |
N |
N |
N |
N |
L |
L |
a? |
|
Ortak değişken hipogamaglobulinemi (çocuk-adult) |
N |
N |
N |
N |
L |
L |
Hiçbiri |
|
A: yok; a: otozomal; H: yüksek; L: düşük;
N: normal; U; bilinmiyor; V: değişken; x: x’e bağlı |
|
|
|
T hücre bozuklukları
T hücre bozuklukları hem hücresel hem de hümoral bağışıklığı
etkileyerek hastaları fungal, protozoal ve viral enfeksiyonlara karşı
duyarlı hale getirir. Sitomegalovirus ve zayıflatılmış kızamık aşısının
neden olduğu viral enfeksiyonlar bu hastalarda ölümcül olabilir.
DiGeorge's Sendromu (Delesyon 22
Sendromu)
En iyi tanımlanmış tanımlanan T hüce defekti konjenital timus aplazi/hipoplazi
olarak da bilinen, immün yetmezliğe hipoparatiroidizmin eşlik ettiği
bir sendromdur. Hipoparatiroidizm, konjenital kalp hastalığı, düşük
kulaklar ve balık ağzının eşlik ettiği bir sendromdur. Bu defektler
paratiroid, timus, dudaklar, kulaklar ve aortik arkın oluştuğu (3.
ve 4. faringeal cep) gebeliğin 6-10. haftalarında fetusun anormal
gelişiminden kaynaklanır. Genetik predispozisyon net değil ve
DiGeorge sendromlu bebeklerin hepsinde timus aplazisi bulunmaz.
Erken fetustan alınan (gestasyonun 13. ve 14. haftaları) timus
grefti tedavide kullanılabilir. Daha büyük greftler GVH reaksiyonu
ile sonuçlanabilir.
Ciddi immün yetmezliği olan DiGeorge hastalarda canlı aşılar aşı
enfeksiyonlarına yol açabilir. Di George sendromu otozomal dominant
kalıtılır ve 22. kromozomdaki delesyon sonucu oluşur (Şekil 2 ve 3).
Delesyonlar değişik boyutlarda görülmesine rağmen hastalık şiddeti
ile arasında korelasyon yoktur. Vakaların yaklaşık % 6’sında
kromozom 22 mikrodelesyonu kalıtsaldır ama çoğu vaka çevresel
faktörlerin sebep olabileceği de novo delesyon sonucu oluşur.
Hastalar timus nakli ile tedavi edilebilir.
|
|
CASE
PRESENTATION
Pediatric
Pathology
DiGeorge Syndrome
A 24-day-old Term Infant with Seizures
(Department of Pathology, University of Pittsburgh) |
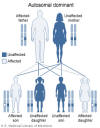 Şekil 2
Şekil 2
DiGeorge sendromunda 22q11.2 delesyonu otozomal dominant patern ile
kalıtılır.
NIH
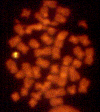 Şekil 3
Şekil 3
Kromozom 22 ‘deki 2 kopyadan sadece birinde floresan işaretleme ile DiGeorge
sendromundaki genlerdeki delesyon gözlemlenebilir.
NIH
|
Değişik
derecelerde B hücre yetmezliği ile birlikte olan T hücre yetmezliği
Ataksia-telanjiektazi
Ataksia-telenjiektazi, yüz bölgesindeki kılcal kan damarlarının
dilatasyonu ve hareket koordinasyonu kaybının (ataksi) eşlik ettiği
T hücre yetmezliğidir. T hücre sayıları ve fonksiyonları farklı
düzeylerde azalmıştır. B hücre sayıları ve IgM düzeyleri normal veya
düşüktür. IgG sıklıkla azalmışken IgA belirgin olarak azalmıştır (vakaların
% 70’ inde). Bu hastalarda başta lösemiler olmak üzere yüksek bir
malignite insidansı vardır. Bu lezyonlar kromozom 14’ te TCR ve
immunglobulin ağır zincir genlerinin bulunduğu bölgede kırımalardan
kaynaklanır. Wiskott-Aldrich
syndrome
Wiskott- Aldrich sendromu normal T hücre sayıları ile ilerleyici
olarak bozulan T hücre fonksiyon azalması ile karakterizedir. IgM
düzeyleri azalmasına rağmen IgG düzeyleri normaldir. IgA ve IgE
düzeyleri artmıştır. Bu sendromun bulunduğu çocuklarda ciddi egzema
ve peteşi (trombositopeni ve platelet defekti sonucu) gelişir.
Polisakkarid antijenlere zayıf cevap verirler ve piyojenik
enfeksiyonlara meyillidirler. Wiskott-Aldrich sendromu X’e bağımlı
kalıtılan bir hücre iskeleti glikoproteini olan CD43‘deki defekt
sonucu oluşur (Şekil 4). MHC
yetmezliği (Çıplak lökosit sendromu)
MHC sınıf 2 transaktivatör (CIITA) proteini kodlayan gendeki
defektin sonucu ASH üzerindeki sınıf 2 MHC moleküllerin kaybı ile
yol açan bir grup immün yetmezlik olgusu tanımlanmıştır. CD4
hücrelerinin timusta pozitif seçilimi MHC moleküllerinin bulunmasına
bağlı olduğundan, bu hastalar daha az CD4 hücresine sahiptir ve
enfeksiyona eğilimlidir. Ayrıca transport ile ilişkili proteini
kodlayan (TAP) gendeki defekte nedeniyle sınıf 1 MHC moleküllerini
eksprese edemeyen ve sonuç olarak CD8+ T hücrelerinde yetmezliğine
sahip bireyler bulunmaktadır.
|
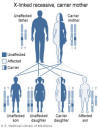 Şekil 4
Şekil 4
Wiskott-Aldrich sendromu X’e bağlı bir hastalıktır
National Library of
Medicine - NIH |
B lenfosit
bozuklukları
T hücre sayılarının ve
fonksiyonlarının normal olduğu bir dizi hastalıkta B hücre sayıları
azalmış veya normal olsa da immunglobulin düzeyleri azalmıştır.
X’e bağlı infantil hipogamaglobulinemi
Bruton hipoglobulinemi veya agamaglobulinemi olarak da bilinen X
geçişli hipogamaglobulinemi, B hücre sayılarının ve tüm
immünglobulin düzeylerinin çok azaldığı en ağır
hipogamaglobulinemidir. Hastalarda defektif B hücresi tirozin kinaz
(btk) geninin yol açtığı B hücresi maturasyon yetersizliği
bulunmaktadır. Bu yüzden B hücreleri ağır zincirleri bulunan fakat
hafif zincirleri yeniden düzenlenmemiş pre-B hücreler olarak
kalırlar. Tanıda immunglobulin ölçümü ve B hücre sayımı kullanılır.
Hastalarda immunglobulinler bulunmaz ve rekürren bakteriel
infeksiyonlara yakalanırlar.
Geçici hipogamaglobulinemi
Çocuklar doğumda anne ile benzer IgG düzeylerine sahiptir. Çünkü IgG’
nin yarı ömrü yaklaşık 30 gündür ve düzeyleri kademeli olarak azalır,
fakat kendi IgG’lerini sentez etmeleri 3 aylıkken başlar. Bazı
infantlarda IgG sentezi 2-3 yaşına kadar başlamayabilir. Bu gecikme
zayıf T hücre yardımına bağlanmaktadır. Bu durum gama globülin ile
tedavi edilebilen geçici IgG yetmezliği ile sonuçlanır.
Yaygın değişken hipogamaglobulinemi (geç başlangıçlı
hipogamaglobulinemi)
Bu bireylerde B hücrelerinin plazma hücrelerine farklılaşmasındaki
başarısızlık nedeniyle 2. ve 3. dekatta IgG ve IgA yetmezliği vardır.
Bu hastalar çeşitli piyojenik bakteri ve intestinal protozoanlara
duyarlıdır. İntravenöz kullanım için özel hazırlanmış gama globülin
ile tedavi edilmelidirler.
IgA yetmezliği
IgA yetmezliği bütün immün yetmezliklerin en yaygın olanıdır (Kafkasyalıların
1/700’ ünde) ve anahtar çevrimindeki bir defektten kaynaklanır. IgA
yetmezliği olan bireylerin yaklaşık % 20’ si düşük IgG’ lere
sahiptir. IgA yetmezliği olan hastalar gastrointestinal, göz ve
nazofarengeal enfeksiyonlara çok duyarlıdır. IgA yetmezliği olan
hastalarda otoimmun hastalık (bilhassa immün kompleks tipi) ve
lenfoid malignansi insidansı yüksektir. Hastaların %30-40’ında anti-IgA
antikorları (IgG yapısında) saptanır ve bu grup gama globülinler ile
tedavi edilmemelidir. Laboratuvar tanısı IgA ölçümü ile yapılır.
Seçici IgG yetmezliği
Farklı IgG alt sınıflarınına ait yetmezlikler bulunmaktadır. Bu
hastalar piyojenik enbfeksiyonlara duyarlıdır.
X’ e bağlı Hiper-IgM İmmünyetmezliği
Bu tip immün yetmezliği olan bireyler düşük IgA ve IgG
konsantrasyonları ile anormal olarak yüksek IgM düzeylerine sahiptir.
Bu hastalar CD4 (+) hücrelerindeki CD40 ligandındaki defekt
nedeniyle IgM’den diğer Ig’lere anahtar çevrimini yapamazlar.
Piyojenik enfeksiyona çok duyarlıdırlar ve intravenöz
gama-globülinle tedavi edilmelidirler.
|
 Şekil 5
Şekil 5
Kronik granülomatoz hastalıkta azalmış intrasellüler bakteri öldürülmesi |
MYELOİD SERİDEKİ NON-SPESİFİK İMMÜN
SİSTEM DEFEKTLERİ Non-spesifik primer immün yetmezlikler
arasında fagositik ve NK hücreler ile kompleman sistemi defektleri
bulunmaktadır.
Konjenital Agranülomatozis
Hastalarda nötrofil sayılarında azalma vardır. Bunun nedeni myeloid
öncül hücrelerin nötrofillere diferansiasyon defektidir. Bu hastalar
granülosit-makrofaj koloni stimulasyon faktörü veya G-CSF ile tedavi
edilirler.
Fagositik sistem defektleri
Fagositik hücrelerin sayı ve/veya fonksiyonlarındaki defektler
çeşitli infeksiyonlara duyarlılıkta artışa yol açabilir.
Siklik nötropeni
Yaklaşık her üç haftada bir dolaşımdaki nötrofil sayılarının azlığı
ile fark edilirler. Nötropeninin sürdüğü yaklaşık bir hafta boyunca
hastalar enfeksiyonlara duyarlıdırlar. Defektin nötrofil
üretimindeki zayıf regülasyonu ile oluştuğu düşünülmektedir.
Kronik granülomatöz hastalık
Kronik granülomatöz hastalık belirgin lenadenopati,
hepatosplenomegali ve lenf nodlarının kronik drenajı ile
karakterizedir. Lökositlerde azalmış intraselüler öldürme ve düşük
respiratuvar patlama vardır (Şekli 5). Bu hastaların çoğunda
yetmezlik NADPH oksidazdaki (sitokrom b558 : gp91phox, veya nadiren
gp22phox) veya fagositoz sonrası respiratuvar patlamada gerekli
diğer kofaktör proteinlerdeki (gp47phox, gp67phox) defektten
kaynaklanır. Bu hastalarda tanı respiratuvar patlamanın ölçüm
yöntemi olan nitroblue tetrazolium (NBT) testinde azalma ile koyulur.
İnterferon gama tedavisi başarılı bulunmuştur.
|
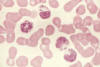 Şekil 6
Şekil 6
Chediak-Higashi Sendromlu hasta preparatında granülosit sitoplazmasında
aşırı büyük granüller görülmekte. Bu görüntü granüllerin oluşumu esmasında
anormal füzyon nedeniyle oluşmaktadır.
Anormal granüller vücutta birçok hücre türünde bulunabilir. |
Lökosit adezyon defekti
Bu hastalıkta T hücreleri ve makrofajlarda, CD11 veya CD18
peptidlerindeki defekt nedeniyle kompleman reseptörü CR3 yoktur ve C3b
opsoninine yanıt veremezler. İlaveten CD11a veya CD11b peptitlerindeki
defek nedeniyle LFA-1 veya MAC-1 gibi integrin moleküllerinde bir defekt
olabilir. Bu moleküller diapedesisde görev alırlar ve bu nedenle
defektif nötrofiller kemotaktik sinyallere etkili cevap veremez.
Tedavisi kemik iliği (MHC uyumlu ve T hücrelerinden arındırılmış)
transplantasyonu veya gen tedavisidir.
Chediak- Higashi sendromu
Chediak-Higashi sendromu azalmış intrasüllüler öldürme ve kemotaktik
hareketin eşlik ettiği fagozom oluşturma, lizozom füzyonu ve proteinaz
yetersizliğinin bulunduğu bir sendromdur. İntraselüler granüller içeren
dev lizozomlar sıklıkla görülür (Şekil 6). Respiratuvar patlama
normaldir. Eşlik eden NK hücre defektleri ile trombosit ve nörolojik
bozukluklar bulunabilir.
|
| |
KOMPLEMAN SİSTEMİ
BOZUKLUKLARI
Kompleman anomalileri de enfeksiyonlara duyarlılıkta artışa
yol açabilir. Kompleman sisteminin farklı komponentlerinin genetik
defektleri vardır, en ciddisi düşük C3 sentezinden veya faktör I ve faktör H
defektinden kaynaklanabilen C3 yetmezliğidir.
|
| |
SEKONDER (KAZANILMIŞ)
İMMÜN YETMEZLİKLER
Enfeksiyonlarla ilişkili immün yetmezlikler
Bakteriel, viral, protozoal, helmintik ve fungal
infeksiyonlar B hücresi, T hücresi, PMNL ve makrofaj yetmezliklerine
yol açabilir. Bunlar arasında en dikkat çekeni kazanılmış
immunyetmezlik sendromudur (AIDS). Sekonder immün yetmezlikler
ayrıca malignitelerde de görülür.
AİDS’teki
immunolojik anormallikler
Kazanılmış immün yetmezliklerin önemli bir oranının
nedeni insan immün yetmezlik virusu (HIV)-1’ in neden olduğu AİDS
tarafından oluşturulmaktadır. İlk olarak 1981 yılında virüs
keşfedildi ve hastalarda Pneumocystis carinii gibi fırsatçı mantar
enfeksiyonları ve Kaposi sarkomu gibi deri tümörleri bulunmaktaydı.
HIV virisünün iki major tipi bulunmaktadır. HIV-1 ve 2. HIV-1
sıklıklıkla kuzey Amerikada bulunmaktadır. Bulaş, cinsel yol,
enfekte kan ve vucüt sıvıları ve anneden fetüse olmaktadır. Bir
retrovirüs olan HIV hücreye girdikten sonra RNA’sını revers
transkriptaz enzimi ile DNA’ya çevirmektedir. DNA konak genomuna
entegre olarak provirüs haline geçer ve hücre ile birlikte replike
olur. HIV-1 diğer çoğu memelide replike olmamasına rağmen
şempanzeleri enfekte edebilmekte fakat AIDS’e dönüşmemektedir. HIV-1
virionu konak hücre kaynaklı lipit çift katmanlı hücre zarından ile
gp41 ve gp120 transmembran glikoproteinlerinden bir zarftan
içermektedir. Gp120 ile CD4 eksprese eden konak hücrelere
bağlanmaktadır. Viral zarfın içinde bulunan viral kor veya
nükleokapsit p17den oluşan matriks proteininden ve içteki p24
proteininden oluşmaktadır. Viral genom iki tek sarmallı RNA molekülü
ile ilişkili revers transkriptaz enzimi ile proteaz ve integraz
enzimlerinden oluşmaktadır.
Replikasyon siklüsü ve
tedavinin hedefleri
Virüs gp120 ile Th hücreler, monosit ve dentritik hücreler
üzerindeki CD4 molekülüne bağlanır. Enfeksiyon için ko-reseptör
gereklidir. Bu ko reseptör molekül, CXCR4 veya CCR5 gibi kemokin
reseptörleridir. CCR5 çoğunlukla makrofajlarda eksprese edilir
ve CXCR4 ise CD4+ T hücrelerde HIV enfeksiyonu için koreseptör
görevi görü. HIV zarfı ile konak hücrenin zarının füzyonu ile
birlikte hükleokapsit hücre içine girer. RT enzimi ile
oluşturulan DNA nükleusa transport edilir ve konak DNA’sı ile
entegre olarak provirüs haline geçer. Provirüs hücre aktive
olana kadar latent olarak kalmaya devam eder. Virionlar
transkribe RNA ve proteinden oluşmaktadır. Hücre membranından
tomurcuklanarak zarf kazanırlar. Bu nedenle terapatik etkenler
viral girişi ve füzyonu hedeflemektedir ve RT enzimini, proteaz
ve integraz enzimini inhibitörlerini içermektedir. Yüksek aktif
anti-retroviral tedavi 3 veya daha fazla bu etken maddeleri
içermektedir.
İmmünolojik
değişikikler
Virüs hızlıca replike olur ve yaklaşık iki hafta içerisinde
hastada ateş ortaya çıkar. Kandaki viral yük anlamlı derecede
artar ve iki ayda pik yaparak sonrasında hızlıca düşer çünkü
lenf nodlarının germinal merkezlerinde latent virüs
bulunmaktadır. Sitotoksik T lenfositler çok erken gelişir ve 3-8
hafta içerisinde antikorlar tespit edilebilir. Yaklaşık 4-8
hafta içerisinde sitotoksik T lenfositlerin Th hücreleri
öldürmesi ile CD4+ T hücre sayısı düşer. Mimetre küpte CD4+ T
hücre sayısı 200’ün altına düştüğünde tam oluşmuş AIDS gelişir.
İmmünoterapi
Etkili bir HIV aşısı geliştirmedeki birçok engel bulunmaktadır.
-
Zayıflatılmış aşı hastalığı uyarabilir
-
CD4+ T hücreler aşı ile yok edilebilir
-
HIV’in birçok antijenik varyantı bulunmakta
-
MHC moleküllerin down regülasyonu ile virüs
düşük immünojeniteye sahiptir
-
Hayvan modeli yoktur
Aşağıdaki reagenler aşı geliştirmede düşünülmektedir:
-
Patojeniteyi düşürmek için mutantların
delesyonu ile immünizasyon
-
Rekombinant proteinlerle aşılama
-
Aşılama için proteinleri kodlayan genler
virüs vektörleri içerinie koyulmaktadır
-
Ko-reseptörlelrle yarışan kemokinler
-
Th hücreleri uyarlak için IL-2 kullanımı
Yaşlanma ile ilişkili immün
yetmezlik
Yaşlanma ile timik kortekste progresif gerileme,
timusun büyüklüğünde gerileme ve hiposellülerite, supresör hücre
fonksiyonunun azalması ve bunun sonucunda otorektivitede artış, CD4
hücre fonksiyonlarında azalış meydana gelmektedir. Zıt olarak B
hücre fonksiyonları bir miktar artmış olabilir.
Malignensi ve
diğer hastalıklarla ilişkili immün yetmezlik
Multiple myloma, Waldenstrom's makroglobulinemisi,
kronik lenfositik lösemi ve iyi diferensiye lenfomada B hücre
yetmezliği bulunmaktadır. Hodgkin's hastalığı ve ilerlemiş solit
tümörler bozuk T hücre fonksiyonu ile ilişkilidir. Malignensinin
tedavisinde kullanılan çoğu terapotik ajan aynı zamanda
immünosüpresiftir.
Sekonder immün yetmezlikler orak hücreli anemi, diabetüs mellitus,
protein kalori malnütrasyonu, yanıklar, alkolik siroz, romatoit
artrit ve renal fonksiyon bozukluğu gibi ibirçok hastalıkta
gelişebilmektedir.
|
|
|
 Mikrobiyoloji ve İmmünoloji On-line, İMMÜNOLOJİ Bölümüne Dönünüz Mikrobiyoloji ve İmmünoloji On-line, İMMÜNOLOJİ Bölümüne Dönünüz
This page last changed on
Saturday, April 02, 2016
Page maintained by
Richard Hunt
|
 Şekil 1
Şekil 1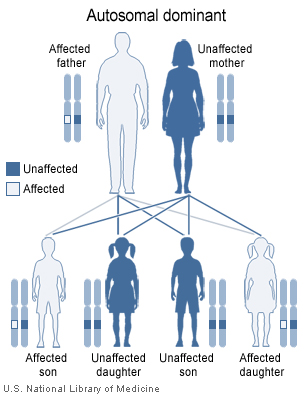 Şekil 2
Şekil 2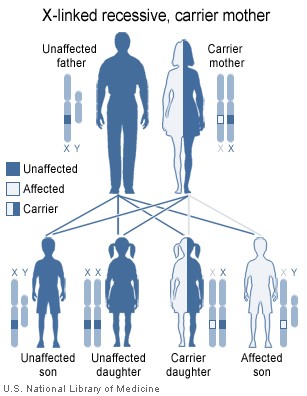 Şekil 4
Şekil 4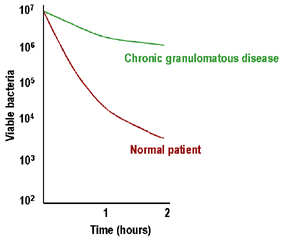 Şekil 5
Şekil 5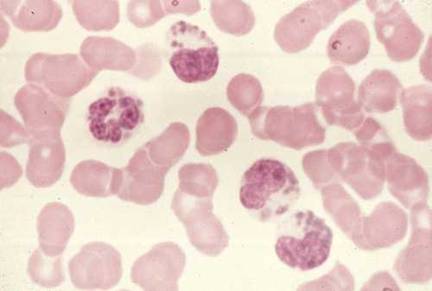 Şekil 6
Şekil 6