|
|
 |
||||||||||||||||||||||||||||||||||
| BACTERIOLOGY | IMMUNOLOGY | MYCOLOGY | PARASITOLOGY | VIROLOGY | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
| Español | |||||||||||||||||||||||||||||||||||
|
Let us know what you think FEEDBACK |
|||||||||||||||||||||||||||||||||||
| SEARCH | |||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||
|
THIS CHAPTER IS IN
SEVERAL PARTS |
|||||||||||||||||||||||||||||||||||
|
WEB RESOURCES Anti-retroviral drugs used in the treatment of HIV infection Approved therapies for the treatment of complications of HIV/AIDS Approved anti-retroviral drugs for pediatric treatment of HIV infection HIV/AIDS Treatment Investigations New Drugs (INDs) Allowed to Proceed |
Anti-HIV chemotherapy has made great strides in the past twenty years from the initial introduction, in 1987, of azidothymidine (AZT) monotherapy (which had little effect on longevity) to the triple drug cocktails that comprise highly active anti-retroviral therapy (HAART) in use today. Although HAART has resulted in major extensions of lifespan and suppression of symptoms in HIV-infected patients, it is not without major side effects. In addition, these drugs penetrate certain parts of the body (e.g. the brain) poorly, are toxic and can select for resistant mutants. Different resistant mutants can emerge in different parts of the body. These drugs target HIV replication and so have no effect on pre-integration complexes or integrated provirus in the latently infected T lymphocytes. As a result, although anti-viral chemotherapy has made HIV infection a tractable disease, it has not led to a cure and new approaches to treating HIV infection are very much needed.
POSSIBLE SITES FOR INTERVENTION IN THE HIV LIFE CYCLE When attempting to develop chemotherapeutic agents to combat viral infections, it is important that the life cycle of the virus be interrupted without killing the host cell. This is difficult because viruses use host cell metabolic process for most of their synthetic reactions. To achieve specificity, processes that are unique to the virus must be identified or, if the agent is toxic to the host cell, it is important that only virus-infected cells be killed. While HIV uses many cellular processes, there are vital functions that are unique to the virus and these have been successfully exploited in drug design. Several stages of the HIV replication cycle are possible targets for chemotherapy:
|
||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||
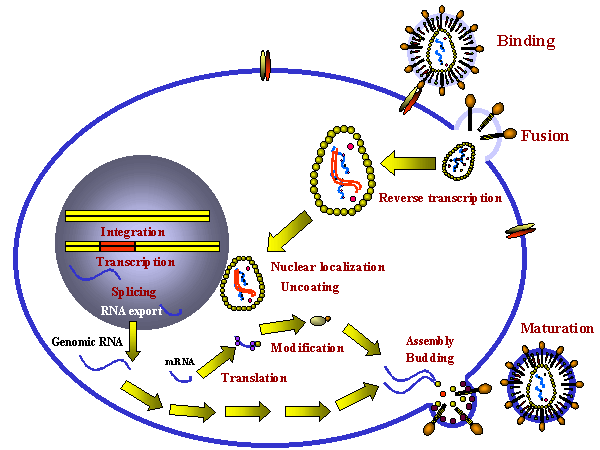 Possible sites for
chemotherapeutic intervention in the HIV life cycle
Possible sites for
chemotherapeutic intervention in the HIV life cycle |
Until recent years, few drugs have shown sufficient activity against HIV at non-toxic concentrations to warrant further development. Effective agents must show activity against virus in T4 cells, macrophages, neural cells, epithelial cells etc. and thereby eliminate virus from the patient. Most agents are virustatic rather than virucidal. This is, perhaps, all we can expect but there are some hopes that a chemotherapeutic approach could lead to the eventual eradication of HIV from the bodies of at least some individuals. However, there are major doubts that eradication can ever be achieved because of the very slow clearance of HIV-infected resting memory T cells. At present, the chemotherapeutic approach that is most likely to eradicate the virus is the Highly Active Anti-Retroviral Therapy (HAART) or triple drug treatment (see below).
|
||||||||||||||||||||||||||||||||||
|
WEB RESOURCES |
|||||||||||||||||||||||||||||||||||
|
INHIBITORS OF TRANSCRIPTION OF THE VIRAL GENOME The single strand HIV RNA genome is first transcribed to double strand DNA by reverse transcriptase, a retroviral-specific enzyme. Since reverse transcription is not a normal function of eukaryotic cells, this is likely to be an important target for anti-HIV chemotherapy and the first anti-HIV drug, azidothymidine (AZT) is such a compound. Nucleoside Reverse Transcriptase Inhibitors These are competitive inhibitors of reverse transcriptase since they bind to the enzyme's active site. In addition, they are DNA chain terminators, that is they are recognized by reverse transcriptase as nucleotides and incorporated into DNA; however, they usually lack the complete deoxyribose ring or a hydroxyl group at carbon 3 of the sugar and therefore their structure precludes the further addition of nucleotides to the nascent polynuceotide since a phosphodiester bond cannot be formed. The first of these competitive reverse transcriptase inhibitors was AZT. Compounds in this category can also be used in cell DNA synthesis leading to chain termination. This is not a problem for cells that are not dividing but is a problem in the replication of erythroid cells, spermatozoa, cell of the immune system and of the gut. Some specificity is achieved because AZT has a far higher affinity for reverse transcriptase that it does for the host cell DNA polymerase but it also inhibits cellular enzymes leading to severe side effects.
|
|||||||||||||||||||||||||||||||||||

Decline in the number of HIV particles in patients treated with a triple drug regimen (HAART). Clinically infected patients were treated with HAART. Plasma viremia declines in two phases: Within 2 weeks, it has declined by 99%. This is followed by a slower decline; by 8 weeks, HIV viral titers have declined below the limits of detection of the assay |
Several reverse transcriptase inhibitors have been approved for use against HIV, none are without side-effects.
|
||||||||||||||||||||||||||||||||||
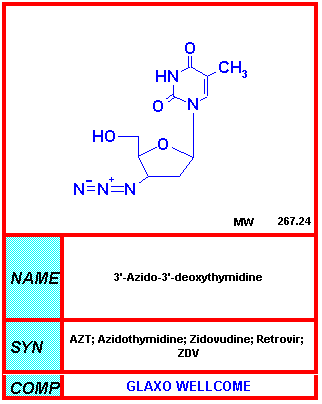 AZT
AZTFrom NIH-NIAID |
Azidothymidine - AZT Zidovudine, Retrovir® AZT is the only nucleoside inhibitor that has been subjected to a placebo-controlled trial. When originally used as the first anti-HIV drug, it was administered when patient CD4 cells were <500 cells/mm3. It delays onset of AIDS-related opportunistic infections but in a controlled trial it was found that the use of AZT in asymptomatic HIV-infected patients resulted in no difference in survival at 3 years. Before the results of a major trial published in April 1993, AZT monotherapy was considered: "Mediocre---but better than nothing". After April 1993, the consensus was that AZT offers "no significant benefit to infected healthy people in slowing the disease or prolonging life" (Concorde trial France/UK. Lancet) When non-syncytium-forming viruses convert to syncytium-forming, there is a change from clinical latency to a rapid decline. Unfortunately, AZT is minimally effective against syncytium-forming version of virus. Nevertheless, AZT is extremely effective at preventing peri-natal transmission of HIV. As a result of the use of AZT monotherapy treatment and similar interventions, the number of perinatally acquired AIDS cases in the United States dropped dramatically. The risk of mother-to-infant transmission of HIV has been found to range from 25% to 35% in an untreated individual. AZT treatment of the mother and her infant can reduce transmission risk from 26% to 8% and AZT/3TC and triple therapy reduce that risk even further. Nevirapine is now the drug of choice for peri-natal treatment (see below). Since AZT also interferes with host cell DNA synthesis, it causes severe side-effects among which are:
AZT can also interfere with other drugs and should not be taken with d4T (stavudine, Zerit, d4T). Moreover, methadone can increase blood levels of AZT, leading to more pronounced side effects. Resistance to AZT occurs rapidly when the drug is used in
monotherapy. |
||||||||||||||||||||||||||||||||||
 From NIH-NIAID
From NIH-NIAID |
Dideoxyinosine - DDI Didanosine, Videx® DDI is a potent inhibitor of reverse transcriptase but is not superior to AZT. It is approved for persons on AZT for > 4 months or who cannot tolerate AZT. Since it is not specific to reverse transcriptase, this drug can also interfere with host cell DNA replication. Among its side effects are:
|
||||||||||||||||||||||||||||||||||
|
Stavudine D4T, Zerit® Stavudine is another reverse transcriptase inhibitor. Again, it has side effects including:
It is used in combination therapy, particularly in advanced HIV disease.
|
|||||||||||||||||||||||||||||||||||
 From NIH-NIAID
From NIH-NIAID |
Didexoycytidine - DDC Hivid®, zalcitabine DDC is the most potent of the three
original nucleoside inhibitors when used on infected cells in culture. It is
approved for use with AZT in adults with <300 CD4 per mm3 who have
significant deterioration.
It is no longer manufactured. |
||||||||||||||||||||||||||||||||||
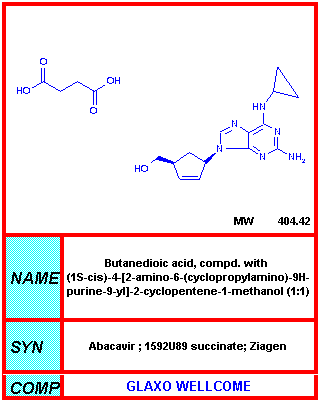 From NIH-NIAID
From NIH-NIAID |
Abacavir Ziagen® Abacavir sulfate (a synthetic
carbocyclic nucleoside) is much less toxic than the above drugs with more
mild side effects such as nausea and headaches Tenofovir Viread®, bis-POC PMPA The main side effects of tenofovir are also nausea and headaches
Why do reverse transcriptase inhibitors not work completely? None of the nucleoside analog reverse transcriptase inhibitors are now used as single drug monotherapy. Instead, they are combined with other drugs as part of HAART. Initially, they may reduce viral burden 50-90% but there is a failure to keep adequate drug levels over long periods of time. Subsequently, resistance develops after six months to one year and occurs more rapidly in patients that have been infected for longer time. This is because they have greater burden of more diverse forms of the virus. Resistance results from mutations in reverse transcriptase which quickly arise because of enormous proliferation rate of virus and the lack of a proof reading activity in host polymerase II. There is thus an enormous section pressure for resistant variants.
|
||||||||||||||||||||||||||||||||||
|
Tenofovir prevents HIV infection in drug
addicts About eight per cent of HIV infections in the United States and ten percent worldwide are caused by needle sharing among drug addicts. In many other countries such as in eastern Europe, the proportion of new infections in drug addicts is much higher. A study involving 2,400 drug users in Thailand, showed that pre-exposure prophylaxis with tenofovir pills reduced the rate of infections by 49 percent. Drug users who took the pills regularly (determined by the level of tenofovir in their blood) did even better since they were 74 percent less likely to become infected. In a similar trial with gay men in the United
States, daily Truvada (Tenofovir and emtricitabine, another nucleoside reverse
transcriptase inhibitor) was found to be 90% effective in preventing infection
in men who took their treatment regularly. |
|||||||||||||||||||||||||||||||||||
|
Non-Nucleoside Inhibitors
The non-nucleoside reverse transcriptase inhibitors are non-competitive inhibitors of the enzyme, that is they bind elsewhere than the active site of the enzyme. Because of the problems with AZT and the other nucleoside analogs in the treatment of HIV, interest grew in another approach to inhibiting the same enzyme, reverse transcriptase. Alternative drugs might be useful in combination therapy since there is a limit to the number of mutations that reverse transcriptase can bear without losing function. Clearly, mutations resistant to a non-nucleoside non-competitive inhibitor of reverse transcriptase would be at a different site in the enzyme from the mutation that makes the enzyme resistant to a competitive nucleoside analog. Thus, although resistant virus strains are cross resistant to other non-nucleoside reverse transcriptase inhibitors, they are not to nucleoside analog inhibitors. Not surprisingly, since these drugs target reverse transcriptase, resistant mutants rapidly emerge, even after only a few passages in cultured cells. In the clinic, resistant mutants also arise rapidly. They are therefore of little use in monotherapy. These are the most potent and selective reverse transcriptase inhibitors that we have, working at nanomolar concentrations. Unlike nucleoside analogs, they have minimal toxicity in tests with cultured cells (anti-viral activity occurs at 10,000 to 100,000-fold lower concentration than the cytotoxic concentration) and they have been shown to work synergistically with nucleoside analogs such as AZT. There is some toxicity when used in humans. These drugs work against nucleoside analog-resistant HIV. Thus, they have high therapeutic index and also show good bioavailablity so that anti-viral concentrations are readily achievable. There is now a collection of such agents that are chemically distinct. The three drugs of this type approved in the U.S. are nevirapine, delavirdine and efavirenz
|
|||||||||||||||||||||||||||||||||||
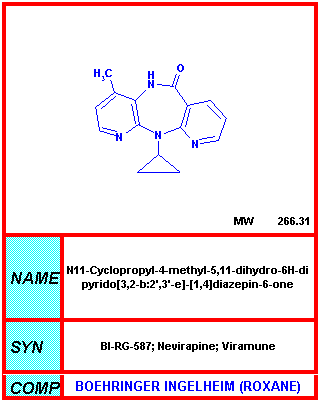 From NIH-NIAID
From NIH-NIAID |
Nevirapine NVP, Viramune®, BIRG-587 In monotherapy, nevirapine gives an initial fall in HIV but resistance sets in and virus titers rise again to a high level. High concentrations that are achieved in the bloodstream may be of some use. A pediatric suspension of nevirapine (Viramune) has been approved by the US Food and Drug Administration for the treatment of HIV-infected infants and children. One side effect of nevirapine is skin rash. This occurs in about one quarter of patients. More serious and much rarer is the skin rash associated with Stevens-Johnson syndrome which can lead to death. In addition, this drug can result in liver damage which in some cases can be fatal. Nevirapine is approved for combination therapy in adults.
|
||||||||||||||||||||||||||||||||||
|
WEB RESOURCES
Mother-to-Infant HIV Transmission Rate Less Than 2 Percent |
|||||||||||||||||||||||||||||||||||
|
Pyridinone derivatives L-697,661 Trials in combination with AZT showed a delay in emergence of resistant virus with these drugs but resistance is NOT prevented. Disappointing results mean that these drugs have not been developed further.
|
|||||||||||||||||||||||||||||||||||
 From NIH-NIAID
From NIH-NIAID |
Delavirdine Rescriptor®, Bis (heteroaryl) piperazine compounds, DLV Delavirdine treatment leads to considerable increases in CD4+ cells in combination therapy (with AZT and 3TC). There have been promising results in patients with very low CD4+ cells that have prior treatment with AZT. In combination with AZT and 3TC, DLV may delay emergence of resistance to AZT. The drug is absorbed rapidly. This drug is used in combination with a nucleoside analog such as AZT and the protease inhibitors discussed below. Skin rash in about one quarter of patients is the major side effect. Atervidine is a similar drug but is no longer in development.
|
||||||||||||||||||||||||||||||||||
|
WEB RESOURCES Rescriptor With Indinavir At Lower Doses Decreases Viral Load |
|||||||||||||||||||||||||||||||||||
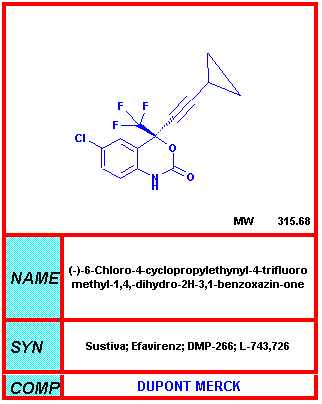 From NIH-NIAID
From NIH-NIAID |
Efavirenz DMP266, Sustiva®, Stocrin® One of the more important treatment developments at the 12th World AIDS Conference was the report of new phase III data showing that Efavirenz (brand name Sustiva), used in combination with other treatment, may suppress viral load at least as well as the protease inhibitor Indinavir in the equivalent combination with nucleoside reverse transcriptase inhibitors. The most noteworthy result was a comparison of viral load reduction with Efavirenz plus AZT plus 3TC, vs. a standard-of-care control group treated with Indinavir plus AZT plus 3TC. The Efavirenz combination suppressed viral load to below 400 copies in a significantly higher proportion of the volunteers than the control arm, at all time points between week 2 and week 24. Side effects include skin rash, nausea, dizziness, diarrhea, headache and insomnia. A small minority of patients develop serious psychiatric problems. In addition, this drug may cause major problems in pregnant women since it is teratogenic in rats. It should not be taken therefore during pregnancy and, especially, in the first trimester.
|
||||||||||||||||||||||||||||||||||
|
Combination Therapy Using Nucleoside Analogs Since the introduction of AZT and similar analogs, new avenues of anti-HIV chemotherapy have been explored since these drugs are clearly not sufficient as anti-HIV agents. This is because of their side effects and, more importantly because of:
AZT has no impact on survival. Combinations of nucleoside inhibitors are more effective in that they reduce plasma HIV RNA to about 10% of the initial value and maintain this for up to 2 years. This can slow development of disease but not prevent it. Mutation rate As always, there remains the problem of mutation of RNA viruses, a phenomenon that results in the rapid evolution of drug-resistant viral quasi species. In an HIV-infected person, about 10.3 x 109 virions are produced per day. These have a half life of 5.7 hours and a fixed mutation rate of 3 x 10-5 per base per replication cycle in an HIV genome of 104 base pairs. This means that at least one mutation may occur in each nucleotide of HIV in a day. After a year from seroconversion, each infected individual establishes his/her own set point of steady state levels of plasma HIV RNA (102 to 107 copies per ml of blood). This level largely determines the rate at which T4 cells are lost. It appears that this level of viral load drives the rate of immune destruction and can predict the course of the disease. Reduction in viral load seems to account for most of the clinical benefits of any drug. Clearly, if viral load is reduced to negligible levels, we have a very important drug. If the negligible levels are achieved by suppression of replication, the evolution of resistant mutants may not be a problem since mutation requires replication.
|
|||||||||||||||||||||||||||||||||||
|
|
Protease inhibitors With the advent of protease inhibitors, there has been optimism that HIV might become a tractable disease since now we have the promise of a drug regimen that can suppress indefinitely the progress of disease. If, as seems to be the case, viral load is the key to the progression from infection to the clinical manifestations of the disease, combination therapy may make HIV infection manageable. Many aspartyl protease inhibitors are
being developed but at present, only a few are approved.
They are all substrate analogs, that is they mimic a peptide that can bind to
the active site of the viral protease. The latter is a dimeric aspartyl protease
(has catalytic aspartates at its active site). When used individually, protease
inhibitors can drive down viral load to between one 30th and one
100th of initial value but sub-optimal doses of these inhibitors
when used alone can result in loss of suppression after several weeks or months
and an accumulation of multiple mutations in the protease gene giving a
high level of drug resistance. However, patients with sustained suppression do
not develop the resistant mutations. This is because replication must be
maintained for the development of such mutations under the selective pressure of
the drug. |
||||||||||||||||||||||||||||||||||
 From NIH-NIAID
From NIH-NIAID |
Saquinavir SQ, Invirase® (there was also a version called Fortovase®) - Made by Hoffman-La Roche This is a hydroxyethylamine transition-state analog of the HIV protease cleavage site. It is the least bio-available of the present protease inhibitors and is the least effective. It is used in combination therapy (HAART). In clinical trials, SQ + AZT + ddC reduced viremia with a rise in T4 cells in individuals with a T4 cell count of 50 - 300/mm3. SQ plus ddC versus any drug alone in individuals with prior AZT treatment showed significant benefit. There are a few side effects in a minority of patients.
These are mild: nausea, diarrhea, upset stomach, and heartburn. |
||||||||||||||||||||||||||||||||||
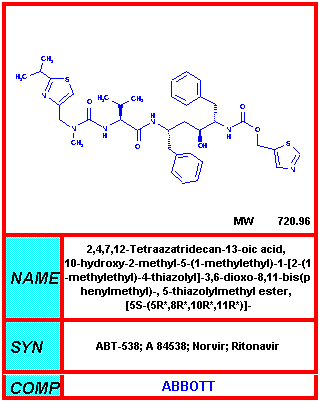 From NIH-NIAID
From NIH-NIAID |
Ritonavir Norvir® - Made by Abbot Labs. Alone Ritonavir reduces AIDS-defining events and death by 58% compared to placebo. This drug is now rarely used alone because of side effects that make it difficult tolerate but it is often used to increase blood levels of other protease inhibitors. This dose much smaller lower than that used in monotherapy or as a single protease inhibitor in HAART and thus causes fewer side effects. Ritonavir causes nausea in 25% of patients. It also causes diarrhea, oral numbness and problems with tasting food. |
||||||||||||||||||||||||||||||||||
 From NIH-NIAID
From NIH-NIAID |
Indinavir Crixivan® - Made by Merke Indinavir is a frequent component of HAART. It plus two anti-reverse transcriptase drugs (AZT and 3TC) reduces HIV to such an extent that PCR cannot detect the virus in 85% of patients. Initial levels in the patients tested were 20,000 to 11,000,000 RNA copies per cu mm of blood (The threshold for detection by PCR is now in the area of 20 copies per ml). This suppression lasted several years in greater than 90% of patients. Indinavir should not be used by pediatric patients or pregnant women. Side effects include kidney stones and anemia. |
||||||||||||||||||||||||||||||||||
|
WEB RESOURCES Crixivan In Triple Therapy Kept HIV Below Detection In Most Patients After Three Years Anti-HIV Combo Of Ziagen+Combivir Equivalent To Crixivan+Combivir At 24 Weeks |
|||||||||||||||||||||||||||||||||||
|
|
HIGHLY ACTIVE ANTI-RETROVIRAL THERAPY (HAART) HAART consists of triple drug therapy. Usually, two nucleoside reverse transcriptase inhibitors are combined with one non-nucleoside reverse transcriptase inhibitor or with one protease inhibitor. For example, one such HAART cocktail consists of zidovudine (AZT) , lamivudine (3TC), both nucleoside analog reverse transcriptase inhibitors, and Indinavir, a protease inhibitor. Another triple drug combination consists of two nucleoside analog reverse transcriptase inhibitors (tenofovir, (R)-9-(2-Phosphonylmethoxypropyl)adenine) and emtricitabine (2',3'-Dideoxy-5-fluoro-3'-thiacytidine) plus the non-nucleoside inhibitors of reverse transcriptase, Efavirenz (Sustiva). The levels of HIV in HAART-treated patients fall to below those in
the blood of long term non-progressors who have remained clinically stable and
shown no loss of T4 cells for over a decade. It was hoped that HAART treatment
might lead to the possibility of purging the patient of the virus as infected
cells eventually died. (This would have to occur in the absence of reinfection by released virus
particles which would be the case if cells only harbored the proviral form of
HIV). It is now known that latent, persistently infected CD4+ cells remain
in the body for long periods. These cells contain the proviral form of the virus
but can reinitiate synthesis of viral particles when reactivated on contact with
antigen. It was formerly the practice to add one additional drug to the treatment as the patient deteriorated. However, this will result in selection of resistant virus and preclude the benefits of combination therapy. If eradication becomes possible, aggressive early treatment is indicated. Although the use of the HAART regimen of drugs can lead to undetectable levels of HIV in the bloodstream, there may still be HIV in genital secretions. Thus these persons are still infective. Recently, patients have been appearing who have clearly been infected by a person who harbors protease inhibitor-resistant virus. This means that patients on the HAART regimen are again having unprotected sex with their partners.
|
||||||||||||||||||||||||||||||||||
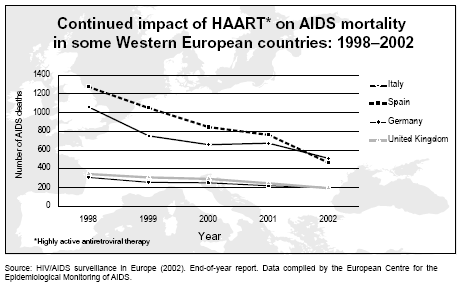 Continued impact of HAART on AIDS mortality
Continued impact of HAART on AIDS mortalityin some Western European countries: 1998–2002. Source: HIV/AIDS surveillance in Europe (2002). End-of-year report. Data compiled by the European Centre for theEpidemiological Monitoring of AIDS. |
At present these drugs must be taken three times a day, every day.. forever, unless they prove to eradicate the virus from the patient. When taken separately, they must be taken under different conditions (empty stomach or after food), making compliance a problem in some patients. Atripla is a combination of three drugs in one pill allowing the HAART regimen as one pill once a day. It contains two nucleoside reverse transcriptase inhibitors, Tenofovir and Emtrictabine ((−)-β-L-3′-thia-2′,3′-dideoxy-5-fluorocytidine) and the non-nucleoside reverse transcriptase inhibitor Efavirenz. It does not contain AZT. Currently protease inhibitors cost about $7000 per year. The cost of zidovudine/lamivudine/protease inhibitor regimen is $12,000 per year. However, HAART is cost effectiveness when compared to the cost of treating a sick AIDS patient. These drugs are, however, no answer to the worldwide AIDS epidemic. In 2001, Cipla, an Indian pharmaceutical company, offered HAART for US$350 per person per year
|
||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
Despite the spectacular results achieved using HAART, these combination therapies are not without side effects. The protease inhibitors, for example, can lead to abnormal redistribution of body fat, called lipodystrophy (40-60% of patients) which may be quite disfiguring. Lipodystrophy results in loss of sub-cutaneous fat. There is increased abdominal girth, enlargement of the dorsocervical fat pad ("buffalo hump"), enlargement of the breasts and fat accumulation around various organs (visceral fat). Some protease inhibitors also lead to red blood cell destruction (hemolytic anemia) and hemorrhaging.
|
|||||||||||||||||||||||||||||||||||
|
OTHER ANTI-HIV DRUGS Although HAART combination therapy has proved highly effective in suppressing viral loads in infected patients, other avenues of treatment are being explored since HAART is not effective in all patients and because of the high costs of this drug combination. Moreover, virus strains that are resistant to HAART have emerged. Drugs that target the initial attachment of HIV to the cell surface It would be clearly logical to target the receptors for HIV on the cell surface since this is the initial stage of infection and is also the rate-limiting step in infection. The first stage in entry is adhesion probably to a charged molecule such as heparan sulfate proteoglycan but it should be noted that the actual mechanism of initial attachment may depend on the cell type. Little progress has been made here.
|
|||||||||||||||||||||||||||||||||||

Cyanovirin-N |
Drugs that inhibit uptake of the virus The uptake of the virus involves the CD4 antigen, the receptor, and chemokine receptor co-receptors. Blocking the interaction of HIV gp120 with either of these could potentially inhibit infection. Antibodies or antibody derivatives that bind to either receptor or gp120 might be useful but none of these agents below can be taken orally. Similarly, receptor or receptor derivatives could act as decoys that bind to HIV gp120, thereby preventing binding to CD4 antigen or co-receptors on the cell surface.
|
||||||||||||||||||||||||||||||||||
 AMD3100 From NIAID
AMD3100 From NIAID |
Agents that block interaction of gp120 with co-receptors
|
||||||||||||||||||||||||||||||||||
 T20 From NIAID
T20 From NIAID |
Agents that block fusion by interacting with gp41
|
||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||
|
Drugs that inhibit uncoating of the nucleocapsid The process of uncoating is not well understood but appears to involve a cell protein called cyclophilin A which appears to be needed for a productive infection by HIV. This cell protein is actually packaged in the viral capsid. However, in vitro studies show that mutants can evolve that no longer require cyclophilin A. |
|||||||||||||||||||||||||||||||||||
Isentress (Raltegravir)
|
Drugs that inhibit integrase
activity Integration of the viral DNA into the host cell chromosome is essential for the production of new virus particles and this would be an excellent target for a specific anti-viral agent since there is no homologous enzyme in humans. A new class of anti-retroviral drugs targets the integration enzyme, the integrase.
|
||||||||||||||||||||||||||||||||||
|
Drugs that block post-translational modifications The matrix protein is myristoylated and the surface glycoprotein is glycosylated. The addition of these post-translational modifications offers a potential target for drug therapy.
Analogs of myristic acid that block myristoyl coA:Myristoyl transferase such as 4-oxatetradecanoic acid are non-toxic and work well in culture. Clinical efficacy is unknown. Blockers of glycosylation Castanospermine, a naturally occurring alkaloid and inhibitor of glucosidase-I has some effect against HIV in vitro. This has led to the development of analogs such as N-Butyl deoxynojirimycin(butyl-DNJ) and 6-butyl-castanospermine which have more potent activity against HIV
|
|||||||||||||||||||||||||||||||||||
|
Nucleic acid based strategies Several possibilities are being investigated. These include:
However, all have delivery problems.
|
|||||||||||||||||||||||||||||||||||
|
WEB RESOURCES Links to other new chemotherapeutic approaches to treating HIV infection |
Thiol-based
reagents |
||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||