![]()

Dr Richard Hunt
EL VIRUS DE LA INMUNODEFICIENCIA HUMANAQUIMIOTERAPIA ANTI-VIH
APÉNDICE IIITraducido por :
Sarah M. Castillo - Jorge, Clinica Corominas
Santiago, Rep. Dominicana
![]()
ENLACES A OTRAS SECCIONES DE VIH Y SIDA AL FINAL DE ESTAPAGINA
HAGA CLICK EN LOS RECUADROS GRISES PARA VENTANAS RAPIDAS CON MÁS INFORMACIÓN
HAGA CLICK EN LOS RECUADROS AMARILLOS PARA SER REDIRECCIONADO A OTROS CAPÍTULOS
FUENTES EN LA RED
Fármacos anti-retrovirales usados en el tratamiento de las infecciones del VIH
Terapias aprobadas para el tratamiento de las complicaciones del VIH/SIDA
Fármacos anti-retrovirales aprobados para el tratamiento de la infección pediátrica del VIH
Tratamiento del VIH/SIDA Investigación de Nuevas Drogas (INDs)
Criterios actuales del tratamiento del VIHLa quimioterapia anti-VIH ha dado grandes pasos en los últimos veinte años desde su producción inicial, en 1987, de la monoterapia con azidotimidina (AZT) (que tenía muy poco efecto longevo) a los cócteles de drogas triples que comprenden la terapia antiretroviral altamente activa (HAART) que se usa hoy en día. Aunque la HAART ha resultado en una mayor extensión del periodo de vida y de la supresión de los síntomas en los pacientes VIH infectados, aún tiene sus efectos secundarios importantes. Además, estas drogas penetran pobremente a ciertas parte del cuerpo (i.e. el cerebro), son tóxicas y pueden seleccionar mutantes resistentes. Los diferentes mutantes resistentes pueden surgir en diferentes partes del cuerpo. Estas drogas atacan la replicación del VIH y por tanto no tienen efecto en los complejos de pre-integración o en el pro virus integrado en los linfocitos T latentemente infectados. Como resultado, auque la quimioterapia anti-viral ha hecho que la infección del VIH sea una enfermedad tratable, no ha llevado a la curación y todavía se necesita de nuevos abordajes en el tratamiento de la infección del VIH.
POSIBLES SITIOS DE INTERVENCIÓN EN EL CICLO DE VIRAL DEL VIH
Cuando se intentan desarrollar agentes quimioterapéuticos para combatir las infecciones virales, es importante que el ciclo de vida del virus sea interrumpido sin eliminar a la célula huésped. Esto es difícil porque los virus utilizan los procesos metabólicos celulares del huésped para sus reacciones sintéticas. Para lograr especificidad, los procesos que son únicos al virus deben de identificarse o, si el agente es tóxico para la célula, es importante que únicamente las células infectadas por el virus sean eliminadas. Mientras que el VIH usa muchos procesos celulares, hay varias funciones vitales que son únicas al virus y estas han sido exitosamente explotadas en el diseño de drogas. Varios estadios del ciclo de replicación del VIH son probables objetivos de la quimioterapia:
- Unión a receptores de superficie
- Fusión de las membranas viral y celular
- Pérdida de la envoltura de la nucleocápside
- Transcripción inversa del ARN a ADN
- Integración del pro virus de AND en el genoma del huésped
- Replicación (AND polimerasa del huésped)
- Transcripción (ARN polimerasa II del huésped)
- Empalme del ARN en el núcleo de la célula huésped (factores virales y del huésped)
- Traducción de proteínas virales
- Proteolisis de las poli proteínas virales (La proteasa del huésped del Cuerpo de Golgi procesa la gp160, la proteasa viral procesa GAG,POL)
- Glicosilación de la gp160
- Fosforilación de proteínas virales
- Acilación grasa de la GAG
- Ensamblaje del virus en la membrana celular del huésped
- Yemación
Las etapas en letra itálica son posibles objetivos puesto que son específicos del virus
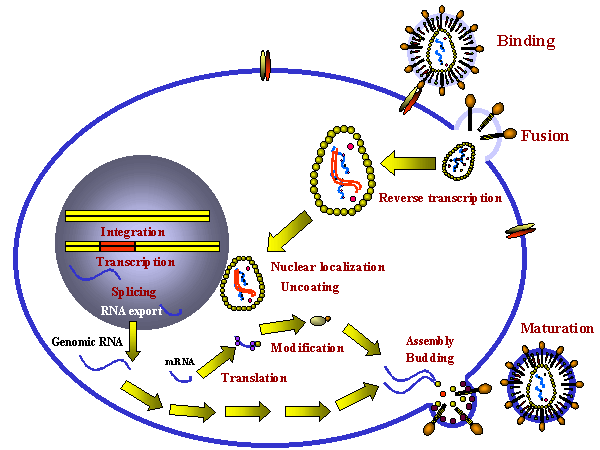 Posibles sitios para intervención quimioterapéutica en el ciclo vital del
VIH
Posibles sitios para intervención quimioterapéutica en el ciclo vital del
VIHHasta años recientes, pocas drogas habían demostrado suficiente actividad contra el VIH a concentraciones no tóxicas como detener un desarrollo posterior. Los agentes efectivos deben mostrar actividad contra el virus en las células T4, macrófagos, células nerviosas, células epiteliales, etc. y así eliminar el virus del paciente.
Muchos de los agentes son virustáticos más que virucidas. Esto es, quizás, todo lo que podemos esperar pero aún hay ilusiones de que un abordaje quimioterapéutico lleve a la eventual erradicación del VIH del organismo de ciertos individuos. No obstante, hay grandes dudas de que la erradicación pueda alcanzarse dada la lenta depuración de las células T de memoria infectadas con el VIH.
En la actualidad, el abordaje quimioterapéutico con más probabilidad de erradicar el virus es la Terapia Anti-Retroviral Altamente Activa (HAART) o tratamiento de triple fármaco (vea más adelante).
FUENTES EN LA RED
INHIBIDORES DE LA TRANSCRIPCIÓN DEL GENOMA VIRAL
El genoma de ARN de cadena sencilla es primero transcribido en una ADN de cadena doble por una transcriptasa inversa, una enzima específica de los retrovirus. Como la transcripción inversa no es una función normal de las células eucariotas, es un objetivo importante de la quimioterapia anti-VIH y la primera droga anti-VIH, la azidotimidina (AZT) actúa a este nivel.
Inhibidores Nucleósidos de la Transcriptasa Inversa
Estos son inhibidores competitivos de la transcriptasa inversa dado que se unen al sitio activo de la enzima. Además, son terminadores de la cadena de ADN, eso es, que son reconocidos por la transcriptasa inversa como nucleótidos y son incorporados al ADN; sin embargo, usualmente carecen del anillo completo de desoxirribosa o de un grupo hidroxilo en el carbón 3 de la cadena carbohidratos y entonces su estructura impide ninguna otra adición de nucleótidos al poli nucleótido naciente porque ya no se puede formar un enlace fosfodiéster. El primero de estos inhibidores competitivos de la transcriptasa inversa fue el AZT. Los compuestos en esta categoría también pueden usarse en la síntesis celular de ADN para finalizar las cadenas. Esto no es un problema para las células que no están en división pero sí para la replicación de los eritrocitos, espermatozoos, células de sistema inmune y de intestino. Se obtiene cierta especificidad porque el AZT tiene una afinidad mucho más grande por la transcriptasa inversa que por la ADN polimerasa del huésped pero también inhibe las enzimas celulares llevando a efectos secundarios severos.

Declinación en el número de partículas de VIH en pacientes tratados con un régimen de drogas triples (HAART). Pacientes clínicamente infectados fueron tratados con HAART. La viremia en plasma declinó en dos fases: En 2 semanas, tuvo una disminución de un 99%. Esto se procede por una disminución más lenta; de 8 semanas, las titulaciones virales del VIH han disminuido por debajo de los límites de detección del ensayo
Varios inhibidores de la transcriptasa inversa han sido aprobados para su uso contra el VIH, pero todos tienen efectos secundarios.
| Inhibidores Nucleósidos de la Transcriptasa Inversa Aprobados | ||
| Droga | Nombre comercial | Enlace |
| AZT (3'-Azidotimidina) | Zidoudine, Retrovir | ZIDOVUDINE - AZT, ZDV (Retrovir) |
| DDI (2',3'-dideoxiinosina) | Didanosine, Videx | DIDANOSINE - VIDEX |
| DDC (2',3'-dideoxicitidina) | Zalcitabine, Hivid | ZALCITABINE (ddC, HIVID®) |
| d4T (Didehidrotimidina) | Stavudine , Zerit | STAVUDINE |
| 3TC (2'-deoxi-3'-tiacitidina) | Lamivudine, Epivir | La FDA Aprueba el Epivir Para su Uso en Pacientes VIH+ de más de 3 meses de edad |
| Succinato de Abacavir | Ziagen | Triple Terapia con Ziagen Efectiva por hasta 48 semanas |
| Fumarato de disoproxil tenofovir | Viread | |
|
Para más información diríjase aquí |
||
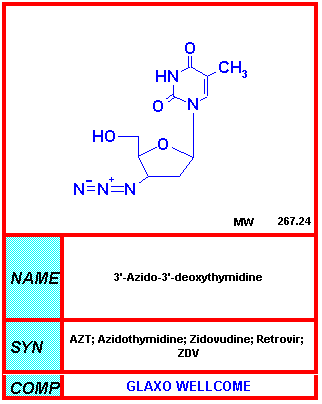 AZT
AZTDel NIH-NIAID
Azidotimidina - AZT
Zidovudine, Retrovir®
La AZT es el único inhibidor que ha sido sujeto de ensayos controlados de placebo. Cuando fue originalmente utilizada como la primera droga anti-VIH, se administraba cuando las células CD4 eran <500 células/mm3. Detiene la instauración de infecciones oportunistas relacionadas al SIDA pero en un ensayo controlado se descubrió que el uso de AZT en pacientes asintomáticos VIH – infectados se encontró que no había diferencia en los sobrevivientes a 3 años.
Antes la publicación de un importante ensayo en Abril de 1993, la monoterapia con AZT era considerada: "Mediocre—pero peor es nada". Luego de Abril de 1993, el consenso será que la AZT ofrecía "ningún beneficio significativo a personas sanas infectadas de retrasar la enfermedad o de prolongar la vida " (Concorde trial France/UK. Lancet)
Cuando los virus no-formadores-de-sincitios se convierten en formadores de sincitios, hay un cambio de latencia clínica a declive rápido. Desafortunadamente, el AZT es mínimamente efectivo contra la versión del virus que formadora de sincitios.
No obstante, el AZT es extremadamente efectivo para prevenir la transmisión peri-natal del VIH. Como resultado del uso del tratamiento del AZT como monoterapia, y de intervenciones similares, el número de casos de SIDA adquirido perinatalmente en los Estados Unidos ha disminuido dramáticamente. El riesgo de transmisión materno-fetal del VIH es de un 25% a un 35% en individuos no tratados. El tratamiento con AZT de la madre y su hijo puede reducir el riesgo de transmisión en un 26% a 8% y AZT/3TC y la terapia triple reducen ese riesgo aún más. La Nevirapina es ahora el fármaco de elección para el tratamiento peri-natal (vea más adelante).
Dado que la AZT también interfiere con la síntesis de ADN de la célula huésped, causa efectos secundarios severos entre los cuales tenemos:
- Anemia (por la inhibición de la síntesis de AND en los precursores de las células eritroides)
- Neutropenia (por la inhibición de la síntesis de AND en la generación de neutrófilos)
- Miopatía (dolor muscular)
La AZT también puede interactuar con otros fármacos y no debería administrarse junto con la d4T (stavudine, Zerit, d4T). Más aún, la metadona puede aumentar los niveles de AZT, llevando a efectos secundarios más pronunciados.
La resistencia a la AZT ocurre rápidamente cuando la droga se usa como monoterapia.
 DDI
DDIDel NIH-NIAID
Dideoxiinosine - DDI
Didanosine, Videx®
El DDI es un potente inhibidor de la transcriptasa inversa pero no es superior a la AZT. Está aprobada para personas tratadas con AZT por > 4 meses o que no puedan tolerar el AZT.
Como no es específica para la transcriptasa inversa, este fármaco también puede interferir con la replicación de AND de la célula huésped. Dentro de sus efectos secundarios tenemos:
- Neuropatía periférica. Esto se da en un quinto de los pacientes y por lo general es temporal si la administración cesa. Podría hacerse permanente si el paciente continua usando el fármaco.
- Pancreatitis. Esta puede ser fatal y ocurre en un 7%
-
Acidosis láctica
Stavudine
D4T, Zerit®
La stavudina es otro inhibidor de la transcriptasa inversa. Dentro de sus efectos secundarios:
- Neuropatía periférica
- Pancreatitis
- Pérdida de leucocitos por fallo de la división celular en la medula ósea
 DDC
DDCDel NIH-NIAID
Didexoicitidina - DDC
Hivid®, zalcitabina
La DDC es el más potente de los tres inhibidores nucleósidos originales cuando se usa en los cultivos de células infectadas. Está aprobada para su uso con AZT en adultos con <300 CD4 por mm3 que tienen un deterioro significativo. Ya no se fabrica.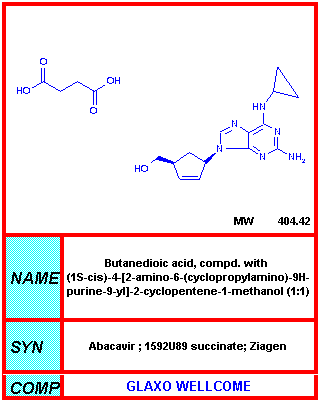
Del NIH-NIAID
Abacavir
Ziagen®
El sulfato de Abacavir (un nucleósido sintético carbocíclico) es mucho menos tóxico que las drogas antes mencionadas con menores efectos secundarios tales como nauseas y cefaleas.
Tenofovir
Viread®, bis-POC PMPA
Los principales efectos colaterales del tenofovir son náuseas y cefaleas
¿Por qué los inhibidores de la transcriptasa inversa no funcionan en su totalidad?
Ninguno de los inhibidores nucleósidos de la transcriptasa inversa se usa en una monoterapia. Al contrario, se combinan con otras drogas como parte de la terapia HAART. Inicialmente, pueden reducir la carga viral en un 50-90% pero se fracasa al querer mantener niveles adecuados del fármaco en largos periodos del tiempo. Subsiguientemente, se desarrolla resistencia luego de seis meses a un año y ocurre más rápidamente en pacientes que han estado infectados por más tiempo. Esto es porque poseen mayor carga viral de más diversas formas del virus. La resistencia es producto de mutaciones en la transcriptasa inversa que rápidamente surgen por la enorme tasa de proliferación del virus y la falta de actividad de corrección de la polimerasa II del huésped. Hay por tanto una enorme sección de presión para las variantes mutantes.
INHIBIDORES No-NucleÓSIDOS
Los inhibidores no nucleósidos de la transcriptasa inversa son inhibidores no competitivos de la enzima, o sea que se unen a otro sitio diferente del sitio activo de la enzima.
Por los problemas del AZT y los otros análogos de nucleósidos en el tratamiento del VIH, creció el interés en otro abordaje para la inhibición de la misma enzima, la transcriptasa inversa. Fármacos alternativos podría ser útiles en terapias de combinación porque hay un límite en el número de mutaciones que aguanta la transcriptasa inversa antes de perder su función. Claramente, las mutaciones resistentes a los inhibidores no nucleósidos no competitivos de la transcriptasa inversa serían en sitios diferentes de los de las mutaciones que hacen la enzima resistente a análogos nucleósidos competitivos. Por tanto, aunque las cepas resistentes del virus manifiestan resistencia cruzada con los otros inhibidores no nucleósidos de la transcriptasa inversa, no la manifiestan con los inhibidores análogos nucleósidos.
No es de sorprender, puesto que estas drogas interfieren con la transcriptasa inversa, que los mutantes resistentes surgen rápidamente, aún después de pocos pasajes en los cultivos celulares. En la clínica, los mutantes resistentes también surgen rápidamente. Por tanto tienen poca utilidad como monoterapia
Estos son los más potentes y selectivos inhibidores de la transcriptasa inversa que se tienen, trabajan a concentraciones nanomolares. A diferencia de los análogos de nucleósidos, tienen mínima toxicidad en los ensayos en cultivos celulares (la actividad anti-viral se ve a concentraciones de 10,000 a 100,000 veces menores que las concentraciones citotóxicas) y han demostrado trabajar sinergísticamente con los análogos nucleósidos como el AZT. Hay cierta toxicidad cuando se emplea en los humanos.
Estos fármacos trabajan contra los análogos nucleósidos resistentes al VIH. Entonces, tienen un alto índice terapéutico y también muestran buena biodisponibilidad de modo que las concentraciones anti-virales son rápidamente alcanzadas.
Ahora hay una colección de dichos agentes que son químicamente distintos. Las tres drogas de este tipo aprobadas en los E. U. son nevirapine, delavirdine y efavirenz.
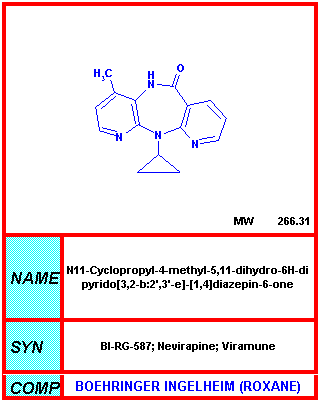
Del NIH-NIAID
Nevirapine
NVP, Viramune®, BIRG-587
En monoterapia, la nevirapine provoca una caída inicial del VIH pero la resistencia se instaura y las titulaciones del virus vuelven a niveles altos. Las altas concentraciones que son alcanzadas en el torrente sanguíneo pueden ser de cierta utilidad. Hay una suspensión pediátrica de nevirapine (Viramune) aprobado por la FDA para el tratamiento de niños e infantes infectados con el VIH.
Un efecto secundario de la nevirapine es un eritema. Ocurre en casi un cuarto de los pacientes. También puede ocurrir un efecto secundario mucho más serio, el Síndrome de Stevens-Johnson que puede llevar a la muerte. Además, esta droga puede provocar daños hepáticos que en algunos casos puede ser fatal.
La Nevirapine está aprobada para terapias de combinación en adultos
|
Quimioterapia e infección infantil de VIH en países en desarrollo |
FUENTES EN
LA RED
NEVIRAPINE
Tasa de transmisión
materno-infantil menos de un 2%
en Fase III del Ensayo Perinatal
Derivados de la Piridinona
L-697,661
Los ensayos en combinación de la AZT mostraron un retraso en el surgimiento de virus resistentes con estas drogas pero la resistencia NO se previene. Los desilusionantes resultados significan que estas drogas no han sido ulteriormente desarrollada.

Del NIH-NIAID
Delavirdine
Rescriptor®, compuestos bis (heteroaril) piperazina, DLV
El tratamiento con delavirdina lleva a aumentos considerables de las células CD4+ en terapia de combinación (con AZT y 3TC). Se han obtenido resultados prometedores en pacientes con muy pocas células CD4+ que han sido tratados previamente con AZT. En combinación con AZT y 3TC, la DLV puede restringir el surgimiento de resistencia a la AZT. La droga se absorbe rápidamente. Esta droga se usa en combinación con un análogo nucleósido como el AZT y un inhibidor de la proteasa (mencionado más adelante).
Los principales efectos adversos son los eritemas de piel que ocurren en un cuarto de los pacientes
La Atervidina es una droga similar pero ya no se fabrica.
FUENTES EN LA RED
Rescriptor con Indinavir en Bajas Dosis Disminuyen la Carga Viral
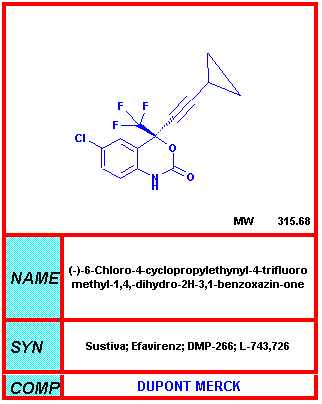
Del NIH-NIAID
Efavirenz
DMP266, Sustiva®, Stocrin®
Uno de los desarrollos más importantes en el tratamiento del SIDA fue reportado en la 12ava. Conferencia Mundial del SIDA de un ensayo en fase III que mostraba que el Efavirenz (nombre comercial Sustiva), usado en combinación con otro tratamiento, podría suprimir la carga viral casi tan bien como el inhibidor de proteasa Indinavir en una combinación equivalente con inhibidores nucleósidos de la transcriptasa inversa. Los resultados más notorios se vieron en una comparación de la reducción de la carga vira con Efavirenz más AZT más 3TC, vs. Un grupo con tratamiento estándar de Indinavir más AZT más 3TC. La combinación del Efavirenz suprimió la carga viral a menos de 400 copias en una proporción significativamente mayor de voluntarios que en el grupo control, en todos los momentos entre las semanas 2 y 24.
Los efectos secundarios incluyen eritema de piel, náusea, mareos, diarrea, cefalea e insomnio. Una minoría de los pacientes desarrolla serios problemas siquiátricos. Además, este fármaco puede causar serios problemas en mujeres embarazadas puesto que es teratogénico en ratas. Debe de administrarse por tanto antes del embarazo y, especialmente, del primer trimestre.
| Inhibidores No-Nucleósidos Aprobados | |
| Droga – Nombre Genérico | Nombres comerciales |
| Delavirdine, DLV | Rescriptor |
| Efavirenz | Sustiva |
| Nevirapine, BI-RG-587 | Viramune |
|
Para más información refiérase aquí |
|
TERAPIA DE CombinaCIÓN UsANDO ANÁLOGOS NUCLEÓSIDOS
Desde la introducción del AZT y análogos similares, se han explorado nuevos caminos de la quimioterapia anti-VIH ya que claramente estas drogas no son suficientes como agentes anti-VIH. Esto se debe a sus efectos secundarios y, más importantemente, a:
- Su habilidad para reducir la carga viral por periodos de tiempo prolongados
- Su habilidad de prolongar la vida
- La alta tasa de evolución de mutantes resistentes en un paciente infectado
La AZT no tiene ningún efecto sobre la supervivencia. Las combinaciones de inhibidores de nucleósidos son más efectivos porque reducen el ARN del VIH en el plasma a más o menos de un 10% del valor inicial y mantienen esto por 2 años. Esto puede endentecer el desarrollo de la enfermedad pero no prevenirla.
Tasa de Mutación
Como ya se ha mencionado, persiste el problema de las mutaciones de los virus de ARN, un fenómeno que provoca la rápida evolución de las casi-especies víricas resistentes a la droga. En personas infectadas por el VIH, se producen cerca de 10.3 x 109 viriones por día. Estas tienen una vida media de 5.7 horas y hay una tasa de mutación fija de 3 x 10-5 por base por ciclo replicativo en un genoma de 104 pares de bases del VIH. Esto significa que por lo menos una mutación por día puede ocurrir en cada nucleótido del VIH.
Luego de un año de la seroconversión, cada individuo infectado establece su propio nivel base estable de ARN del VIH en el plasma (102 a 107 copias por ml de sangre). Este nivel determina en gran medida la tasa a la que se pierden las células T4. Parece que este nivel de carga viral lleva la tasa de destrucción inmune y puede predecir el curso de la enfermedad. La reducción de la carga viral parece ser responsable de los principales beneficios clínicos clínicos de cualquier fármaco. Claramente, si la carga viral se reduce a niveles insignificantes, se tiene un fármaco muy importante. Si se alcanzan estos niveles insignificantes con la supresión de la replicación, la evolución de mutantes resistentes puede no ser un problema ya que la mutación requiere de replicación.
INHIBIDORES DE LA PROTEASA
Con el advenimiento de los inhibidores de la proteasa, ha surgido optimismo de que el VIH pueda volverse una enfermedad tratable ya que ahora tenemos la promesa de un régimen farmacológico que puede suprimir indefinidamente le progreso de la enfermedad. Sí, como parece ser el caso, la carga viral es la clave de la progresión de la infección a las manifestaciones clínicas de la enfermedad, la terapia de combinación puede hacer que la infección del VIH sea manejable.
Muchos inhibidores de la aspartil proteasa están siendo desarrollados pero en la actualidad, sólo unos pocos han sido aprobados. Son todos análogos de sustratos, que simulan un péptido que puede unirse al sitio activo de la proteasa viral. Esta última es un dímero de la aspartil proteasa (tiene aspartatos catalíticos en su sitio activo). Cuando se utilizan individualmente, los inhibidores de la proteasa pueden disminuir la carga viral a menos de una 30ava a una 100ma del valor inicial pero las dosis sub-óptimas de estos inhibidores utilizadas solas resultaban en un pérdida de la supresión luego de varias semanas o meses y la acumulación de múltiples mutaciones en el gen de la proteasa proveyendo un alto nivel de resistencia farmacológica. No obstante, los pacientes son una supresión sostenida no desarrollan las mutaciones resistentes. Esto es porque la replicación debe mantenerse para el desarrollo de tales mutaciones bajo la presión selectiva del fármaco.
Del NIH-NIAID
Saquinavir
SQ, Invirase® (también hubo una versión llamada Fortovase®) – Hecha por Hoffman-La Roche
Este es un análogo de estado de transición hidroxietilamina del sitio de segmentación de la proteasa del VIH. Es el menos biodisponible de los actuales inhibidores de la proteasa y es el menos efectivo. Se utiliza en terapias de combinación (HAART). En ensayos clínicos, SQ + AZT + ddC redujeron la viremia con un aumento de las células T4 en individuos con un conteo de células T4 de 50 - 300/mm3. SQ más ddC versus cualquier droga sola en individuos con un tratamiento previo de AZT demostraron un beneficio significativo.
Hay algunos efectos secundarios en una minoría de los pacientes. Estos son leves: nausea, diarrea, malestar estomacal, y acidez.
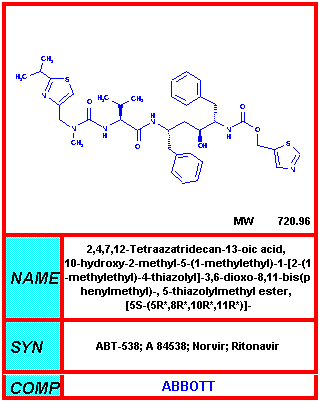
Del NIH-NIAID
Ritonavir
Norvir® - Fabricado por Laboratorios Abbot.
Sólo el Ritonavir reduce los eventos que definen el SIDA y la muerte en un 50% comparado al placebo.
Hoy en día, esta droga rara vez se usa sola porque sus efectos secundarios la hacen difícil de tolerar pero con frecuencia se usa para aumentar los niveles sanguíneos de otros inhibidores de la proteasa. Esta dosis mucho menor que la usada en la monoterapia o como el único inhibidor de la proteasa en una HAART y por tanto tiene menos efectos secundarios. El Ritonavir causa nausea en el 25% de los pacientes. También causa diarrea, parestesia oral y problemas de la sensación del gusto.

Del NIH-NIAID
Indinavir
Crixivan® - Hecho por Merke
El Indinavir es un componente frecuente de la terapia HAART. Junto a dos fármacos anti – transcriptasa inversa (AZT y 3TC) reduce el VIH de tal modo que la PCR no puede detectar el virus en un 85% de los pacientes. Los niveles iniciales analizados en los pacientes fueron de 20,000 a 11, 000,000 de copias de ARN por mm cúbico de sangre (El umbral para la detección por PCR es ahora alrededor de 20 copias por ml). Esta supresión permanecía varios años en más del 90% de los pacientes.
El Indinavir no debería usarse en pacientes pediátricos o en mujeres embarazadas. Los efectos secundarios incluyen litiasis renal y anemia.
FUENTES EN LA RED
Combo anti-VIH de Ziagen+Combivir Equivalente a Crixivan+Combivir a las 24 semanas
TERAPIA ANTIRETROVIRAL ALTAMENTE ACTIVA (HAART)
La HAART consiste en un terapia triple de fármacos. Generalmente, dos inhibidores nucleósidos de la transcriptasa inversa se combinan con un inhibidor no nucleósido de la transcriptasa inversa o con un inhibidor de proteasa. Por ejemplo, uno de estos cócteles HAART consiste en zidovudine (AZT) , lamivudine (3TC), ambos análogos nucleósidos de la transcriptasa inversa, e Indinavir, un inhibidor de la proteasa. Otra combinación triple consiste en dos análogos nucleótidos de la transcriptasa inversa (tenofovir, (R)-9-(2-Fosfonilmetoxipropil) adenina) y emtricitabine (2',3'-Dideoxi-5-fluoro-3'-tiacitidina) más un inhibidor no nucleósido de la transcriptasa inversa, el Efavirenz (Sustiva).
Los niveles de VIH en los pacientes tratados con HAART caen por debajo de los de aquellos que son no-progresores de larga data que han permanecido clínicamente estables y no han mostrado pérdida de células T4 en más de un década. Se esperaba que el tratamiento HAART llevara a la posibilidad de purgar el paciente del virus a medida que se eliminaban las células infectadas. (Esto hubiese ocurrido en la ausencia de reinfección por la liberación de partículas víricas que es el caso si las células solo albergaran la forma pro viral del VIH). Actualmente se conoce que células CD4+ latentes, persistentemente infectadas permanecen el cuerpo por largos periodos. Estas células contienen la forma pro viral del virus pero pueden reiniciar su síntesis cuando son reactivadas al contacto con el antígeno.|
¿Podría la terapia HAART ser una cura para la infección del VIH? |
Anteriormente su usaba el añadir un fármaco adicional al tratamiento del paciente en deterioro. No obstante, esto provocaría la selección de un virus resistente e impediría los beneficios de la terapia de combinación. Si la erradicación se hace posible, un tratamiento temprano agresivo es lo recomendado.
Aunque el uso del régimen HAART puede llevar a niveles indetectables de VIH en la sangre, aún puede haber IH en las secreciones genitales. Entonces estas personas aún son infecciosas. Recientemente, han surgido pacientes que claramente habían sido infectados por personas que portaba el virus resistente a inhibidor de proteasa. Esto significa que los pacientes bajo el régimen HAART estaban teniendo relaciones sin protección con sus parejas.
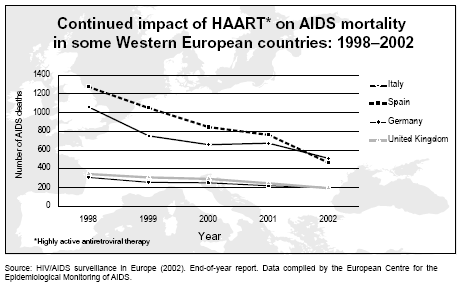 Impacto continuado de la HAART en la mortalidad del sida en algunos países
de Europa occidental: 1998–2002. Fuente: Sondeo del VIH/SIDA en Europa
(2002). Reporte de fin de año. Data compilada del Centro Europeo para el
monitoreo epidemiológico del SIDA.
Impacto continuado de la HAART en la mortalidad del sida en algunos países
de Europa occidental: 1998–2002. Fuente: Sondeo del VIH/SIDA en Europa
(2002). Reporte de fin de año. Data compilada del Centro Europeo para el
monitoreo epidemiológico del SIDA.En la actualidad estas drogas pueden ser tomadas tres veces al día, una vez al día… por todo lo que resta de vida, a menos que prueben que erradican el virus del paciente. Cuando se toman por separado, deben de administrarse bajo condiciones diferentes (con el estómago vacío o después de las comidas), lo que hace que la complianza sea problemática para algunos pacientes. La Atripla es una combinación de las tres drogas en una sola pastilla permitiendo que el régimen HAART sea de una pastilla una vez al día. Contiene dos inhibidores nucleósidos de la transcriptasa inversa, Tenofovir y Emtrictabine ((−)-β-L-3′-tia-2′,3′-dideoxi-5-fluorocitidina) y un inhibidor no nucleósido de la transcriptasa inversa, Efavirenz. No contiene AZT.
Actualmente los inhibidores de la proteasa cuestan cerca de US$7000 al año. El costo del régimen zidovudine/lamivudine/inhibidor de proteasa es de US$12,000 por año. Sin embargo, la HAART es costo efectiva, cuando se compara el costo del tratamiento de un paciente con SIDA manifiesto. Estos fármacos, no obstante, no son la repuesta a la epidemia mundial del SIDA. En el 2001, Cipla, una compañía farmacéutica de la India, ofreció la HAART por US$350 por persona por año
|
Criterios del tratamiento HAART de pacientes sin exposición previa a terapia anti-VIH |
| Inhibidores de la Proteasa Aprobados | |
| Fármaco | Nombre comercial |
| Saquinavir | Fortonase |
| Mesilato de Saquinavir, SQV | Invirase |
| Ritonavir, ABT-538 | Norvir |
| lopinavir y Ritonavir | Kaletra |
| Indinavir, IDV, MK-639 | Crixivan |
| Mesilato de Nelfinavir, NFV | Viracept |
| Amprenavir | Agenerase |
| Fosamprenavir Calcium | Lexiva |
| Tipranavir | Aptivus |
| Sulfato de Atazanavir | Reyataz |
Efectos secundarios de la HAART
A pesar de los espectaculares resultados alcanzados usando la HAART, estas terapias de combinación tienen sus efectos secundarios. Los inhibidores de la proteasa, por ejemplo, pueden provocar una redistribución anormal de la grasa corporal, llamada lipodistrofia (40-60% de los pacientes) que puede ser bastante desfigurante. La lipodistrofia es producto de la pérdida la grasa subcutánea. Hay un aumento en las medidas abdominales, agrandamiento de la grasa dorso cervical ("joroba de búfalo"), agrandamiento de las glándulas mamarias y acumulación de grasa alrededor de varios órganos (grasa visceral). Algunos inhibidores de la proteasa también han llevado a la destrucción de glóbulos rojos (anemia hemolítica) y hemorragias.
OTROS FÁRMACOS ANTI-VIH
Aunque la terapia de combinación HAART ha probado ser altamente efectiva en la supresión de las cargas virales en pacientes infectados, se han explorado otras vías del tratamiento puesto que la HAART no es efectiva en todos los pacientes y por el alto costo de este combinación farmacológica. Más aún, ya hay cepas que son resistentes a la HAART.
Fármacos que atacan la fijación inicial del VIH a la superficie celular
Sería lo más lógico atacar a los receptores del VIH en la superficie celular dado que esta es la fase inicial de la infección y también el paso limitante en la infección. La primera etapa de la entrada es la fijación a una molécula probablemente cargada como los proteglicanos de heparán sulfato pero deberíamos hacer notar que el mecanismo actual de la fijación inicial puede depender del tipo de célula. Se ha alcanzado muy poco progreso en esta área.

Cianovirin-N
Fármacos que inhiben la absorción del virus
La absorción del virus implica un antígeno CD4, el receptor, y un receptor de quimiocinas como co-receptor. Bloqueando la interacción de la gp120 del VIH con cualquiera de estas podría potencialmente inhibir la infección. Los agentes que se unen al receptor o a la gp120 pueden ser útiles pero ninguno de los agentes que se mencionan más adelante puede ser administrados por vía oral.
CD4 Soluble
Una preparación de la región de la molécula CD4 que se une a la gp120, bloquearía la fijación del CD4 en la superficie celular y el CD4 soluble bloquea la infección en los cultivos. Desafortunadamente, tiene muy pocos efectos clínicos. Este abordaje no es muy práctico porque la proteína se degrada muy rápidamente. Además, puede exacerbar la infectividad ya que promueve la unión del VIH a los receptores de quimiocinas.CD4-Ig2 (PRO542)
El PRO 542 es una forma tetramérica del antígeno CD4 soluble genéticamente fundido a una inmunoglobulina. Esta proteínas de fusión CD4-inmunoglobulina comprende los dominios D1 y D2 del CD4 humano y las cadenas pesadas y ligeras de las regiones constantes del la IgG2 humana. Tiene alta afinidad por la gp120.CD4-PEG (endotoxina A de la Pseudomonas aeruginosa)
Este fármaco sinergiza con la AZT pero puede tener una toxicidad no específica muy grandeAnticuerpos que se unen al antígeno CD4
Los anticuerpos que se unen al antígeno CD4 probablemente bloquean la fijación del virus pero podría ser inmunosupresores porque implicaría una depleción de las células CD4.
 AMD3100
AMD3100
Del NIH-NIAID
Agentes que bloquean la interacción de la gp120 con co-receptores
Antagonistas CCR5
El papel de los co-receptores fue descubierto cuando se demostró que algunas quimiocinas bloqueaban la infección. El CCR5 es un co-receptor del VIH y las deleciones del CCR5 hacen que el paciente infectado sea resistente al VIH. Las personas con esta deleción parecen ser normales y sanos. Esto sugiere que atacar al CCR5 puede bloquear el VIH en los pacientes sin tener ningún efecto severo en el sistema inmunológico. La RANTES es una quimiocina que se une al CCR5 y a un análogo del RANTES (n-nonanoil (NNY)-RANTES) que se ha demostrado que bloquea la infección del VIH sin disparar ningún señal intracelular mesurable del receptor. No obstante, aunque los antagonistas del co-receptor CCR5 son efectivos para inhibir la replicación del VIH no-inductor-de-sincitio, estos proveen una presión selectiva para la producción de cepas víricas inductoras de sincitio (X4) las cuales son resistentes al análogo de la RANTES y pueden usar el co-receptor CXCR4. Más aún, los derivados de las quimiocinas son proteínas (o por lo menos, péptidos) y por ellos no están disponibles para administración oral y probablemente tengas vidas medias cortas in vivo. Los anticuerpos monoclonales contra el co-receptor pueden ser mejores y trabajar efectivamente en el bloqueo de la infección in vitro pero tampoco estarán disponibles para administración por vía ora.
Se han descubierto antagonistas de los co-receptores constituidos de pequeñas moléculas. Estos con frecuencia tienen una alta carga negativa. Además, los péptidos catiónicos derivados de la secuencia V3 de la gp120 parecen funcionar. Debido a su carga, los antagonistas pequeños de los co-receptores tiene muy poca biodisponibilidad oral pero el AMD3100 está bajo ensayos clínicos en los cuales se administra mediante inyección. Parece que se une al CXCR4 (fusina) y bloquea la interacción entre el CXCR4 y la V3.
 T20
T20
Del NIH-NIAID
Agentes que bloquean la fusión al interactuar con la gp41
Fuzeon
Péptidos derivados de la gp41 pueden inhibir la infección, probablemente bloqueando la interacción de la gp41 con las proteínas de membrana celular durante la fusión o impidiendo el cambio conformacional que resulta de la asociación de los moléculas de gp41 que es necesario para la fusión. El T-20 (Fuzeon) bloquea este cambio conformacional. En ensayos clínicos, se logró una reducción de casi dos log en la reducción de los niveles virales plasmáticos. Esta droga fue aprobada en el 2003 pero los reportes recientes sugieren que tiene baja biodisponibilidad y causan el surgimiento de mutantes resistentes.
Hay una hendidura en la gp41 que pudiera albergar un inhibidor pequeño. Los péptidos que contienen D-aminoácidos que podría entrar en esta cavidad han sido identificados e inhiben la fusión.
|
Inhibidores de Fusión Aprobados |
|
|
Droga |
Nombre Comercial |
|
Enfuvirtide, EFV, T-20 |
Fuzeon |
|
|
|
Cianovirin
La Cianovirin-N (CV-N) es una proteína de las cianobacterias (algas verde-azules) Nostoc ellipsosporum y actúa como un inhibidor de la fusión del VIH mostrando actividad tanto contra el VIH-1 como el VIH-2 in vitro y en animales.
Fármacos que inhiben la pérdida de la envoltura de la nucleocápside
El proceso de pérdida de la envoltura de la nucleocápside aún no está esclarecido del todo pero parece implicar una proteína celular llamada ciclofilina A, la cual parece ser necesaria durante una producción productiva del VIH. Esta proteína celular por lo general, es empacada en la cápside viral. No obstante, los estudios in vitro muestran que pueden surgir mutantes que ya no requieran de la ciclofilina A.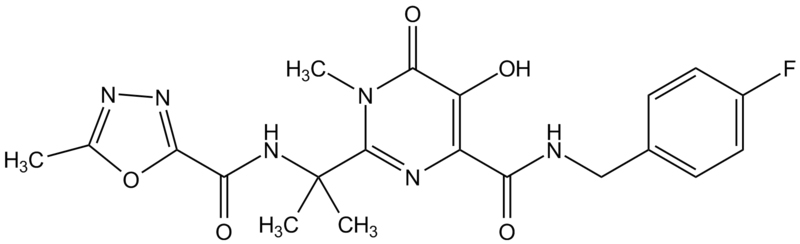
Isentress (Raltegravir)
Ácido L-chicoric
Ácido dicafeoilquinico
Fármacos que inhiben la actividad de la integrasa
La integración del ADN viral al cromosoma de la célula huésped es esencial para la producción de nuevas partículas virales y esto sería un excelente paso para intervenir con agentes antivirales específicos puesto que no hay ninguna enzima homóloga en los humanos. Una nueva clase de drogas anti-retrovirales atacan la enzima de integración, la integrasa.
Isentress® (Raltegravir, MK-0518)
El Isentress fue aprobado para su uso en adultos por la USFDA en Octubre de 2007. Se puede usar como parte del régimen HAART cuando el paciente es resistente a otras drogas tales como los inhibidores de proteasa. Se comparó al Sustiva (terapia de elección) en HAART por un periodo de más de 24 semanas. Más del 80 porciento de aquellos que tomaron el fármaco manifestaron una reducción en los niveles hemáticos del virus hasta niveles apenas detectables. No está aprobado para niños con VIH.El Zintevir (AR177) es un oligonucleótidos de 17 bases con dos cuartetos de deoxiguanosinas y se creyó que era un inhibidor de la integrasa. Ya ha sido sometido a ensayos clínicos de fase I y II.
El ácido L-Chicórico previene la replicación del VIH-1 mediante la inhibición de la VIH-1 integrasa. El 3,5-DCQA, un ácido dicafeoilquínico, aislado de Baccharis genistelloides, es también un potente inhibidor de la VIH-1 integrasa y se une al sitio activo de la enzima.
Drogas que bloquean las modificaciones post-traduccionales
La proteína de matriz es miristoilada y la glicoproteína de superficie es glicosilada. La adición de estas modificaciones post-traduccionales ofrecen un blanco potencial para la terapia farmacológica.
Bloqueadores de la miristoilación
Los análogos del ácido mirístico que bloquean la miristoil coA:Miristoil transferasa tales como el ácido 4-oxatetradecanoico no son tóxicos y trabajan bien en cultivos. La eficacia clínica aún es desconocida.Bloqueadores de la glicosilación
La castanospermina, un alcaloide natural e inhibidor de la glucosidasa-I tiene ciertos efectos in vitro contra el VIH. Esto ha llevado al desarrollo de análogos como el N-Butil deoxinojirimicin(butil-DNJ) y la 6-butil-castanospermina que tienen un efecto más potente contra el VIH.
Estrategias basadas en los ácidos nucleicos
Hay varias posibilidades que están siendo estudiadas. Estas incluyen:
· ARN de Anti-sentido que se unen a los ARNm del VIH y bloquean su traducción
· ARN Catalíticos (ribozimes)
· Oligonucleótidos que atacan las proteínas de control de unión a ácidos nucleicos tales como las TAT y REV.
Sin embargo, todas han derivado algún problema.
FUENTES EN LA RED
Enlaces a otros abordajes quimoterapéuticos nuevos para el tratamiento de la infección del VIH
Reactivos con compuestos tiol
La N-acetil cisteína y la procisteína pueden inactivar el VIH en células
latentemente infectadas en las que aumentan el glutatión (GSH) intracelular.
Los 1,2-ditiol-3-tiones aumentan el GSH e inhiben la transcriptasa inversa
de modo irreversible.
![]() Regresso a Parte 1
Regresso a Parte 1
![]() Regreso a la sección de
Virologia
Microbiología e Inmunología on line
Regreso a la sección de
Virologia
Microbiología e Inmunología on line
Derechos de autor 2007 The Board of Trustees of the University of South Carolina
Esta página se modificó
recientemente en
Wednesday, July 30, 2008
Mantenimiento de la pagina por
Richard Hunt
Favor de reportar problemas a
rhunt@med.sc.edu
OTRAS SECCIONES SOBRE EL VIH
PARTE I VIRUS DE LA INMUNODEFICIENCIA HUMANA Y EL SIDA
PARTE II VIH Y SIDA, LA ENFERMEDAD
PARTE III CURSO CLÍNICO DE LA ENFERMEDAD
PARTE IV PROGRESIÓN Y COFACTORES
PARTE VI SUBTIPOS Y CO-RECEPTORES
PARTE VII COMPONENTES Y CICLO DE VIDA DEL VIH
PARTE X PERDIDA DE CÉLULAS CD4
PARTE XI OTRAS CÉLULAS INFECTADAS POR EL VIH Y POLIMORFISMOS POBLACIONALES
APÉNDICE II ¿ES EL VIH CAUSA DE SIDA?
APÉNDICE III QUIMIOTERAPIA ANTI-VIH