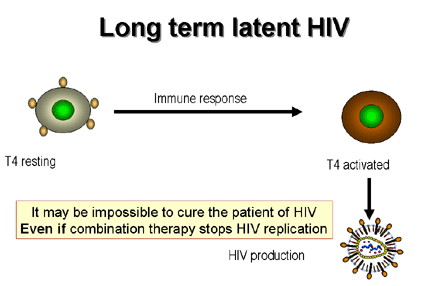
Figure 21
Figure 21
Dynamics of CD4 cells in HIV infection.
 Figure 22
Figure 22
Activation of infected T4 cells
 Figure 23
Figure 23
Latency |
CELLULAR LATENCY
After T4 cells have been activated and
proliferate in an immune response to a particular antigen, most die by apoptosis
as the response to that antigen wanes. However, some of the cells do not
die and become dormant (they are sometimes referred to as resting T cells). They remain for a long time in the body
so that they can respond promptly to a second exposure to the same antigen. This
is called immunological memory and is why the response to the second encounter
with an antigen is more rapid than the primary response. The T4 cells that
become dormant are called memory T cells and can remain quiescent
(non-proliferating) for decades.
If an activated T4 cell happens to be infected
by HIV, it also is most likely to die by apoptosis but a few of these cells become memory cells. In their quiescent state they do not replicate the
virus but still harbor it as a DNA copy integrated into the chromosomes, the
provirus (figure 21). On immune stimulation of the infected memory cells, not
only are genes expressed that are important in the immune response but also HIV
genes are expressed leading to production of new virus particles (figure 22).
The dormancy of the virus in resting memory
T cells is referred to as cellular
latency and may last for a few hours
or days or very much longer in a small minority of cells. It was hoped that the used of highly active anti-retroviral therapies (HAART)
would eliminate the virus from the patient altogether but the memory T4 cells may provide a reservoir of integrated virus that cannot be
eliminated by chemotherapy and may persist for a lifetime.
Latency is broken when the virus
starts to proliferate
and this occurs when the T4 cell is stimulated during an antigenic response.
MECHANISM OF CELLULAR LATENCY
HIV mostly infects cells that express
CD4 antigen and the appropriate co-receptors. This means that T4 lymphocytes are
the primary target of the virus. However, the virus can only replicate in
activated (actively dividing) cells but not in infected naïve T4 cells (those
that have not been activated by antigen) or in resting memory T4 cells (those
that have been activated but have not undergone apoptosis and have returned to
the quiescent state). In these cells the virus is said to be latent.
In the naïve cells, most HIV is not integrated into the chromosomes. The virus
has been reversed transcribed but remains as a provirus (the DNA form of the
virus) in the cytoplasm. In memory T4 cells, the provirus has integrated into
the host cell chromosomes where it is usually found in the introns of actively
transcribed genes. Here, it remains in a latent state until the cell is
reactivated on contact with antigen.
So why is the provirus only replicated in activated T4 cells? There have been
several suggestions:
-
Quiescent T4 cells may lack
sufficient small molecules (for example, nucleotides) needed to make RNA.
-
For complete transcription of the
HIV genome into RNA, the presence of a small viral protein, Trans-Activator
of Transcription (TAT) is required. In the absence of TAT, transcription
terminates prematurely.
-
There could also be a lack of
necessary host cell-provided transcription factors in resting T4 cells or
the presence of host cell-encoded transcription terminators.
-
The integrated provirus may not be
accessible to the transcription machinery of the resting cell. This would
seem unlikely, however, since, as noted above, proviruses are often found in the introns of
actively transcribed genes.
Possibly, the provirus is continually
transcribed in memory cells but the viral RNA cannot get out of the nucleus
because of the need for splicing prior to export. Normally, nuclear export of
unspliced HIV RNA depends on expression of another small HIV encoded protein
called REV (Regulator of Virion protein expression) which may be lacking in
quiescent T4 cells.
Two forms of HIV cellular latency have been suggested. In pre-integration
latency, the virus enters the naïve resting cell but, after reverse
transcription, mostly remains in the cytoplasm as a full length provirus,
probably because the low ATP levels preclude the energy-dependent import of the
pre-integration complex. ATP levels rise when a T4 cell is activated and nuclear
import is followed by integration and transcription. Some HIV may enter the
nucleus of the naïve cell but it is only slowly transcribed because the
necessary nucleotides are in short supply. Many of these transcripts are never
completed and are degraded by the cell. Pre-integration latency is probably not
very important clinically.
Post-integration latency is clinically very important since it is characteristic
of long lived memory T4 cells. These are the cells that have been activated and
reverted to the resting state. Again, little virus is produced in these cells
even though the virus integrates into the introns of genes that are actively
transcribed. It had been thought that HIV might integrate into regions of the
chromosomes that are preferentially repressed in resting cells but this seems
not to be the case. Lack of HIV transcription might result from the phenomenon
of transcriptional interference in which the transcription of an active gene is
initiated at its promoter and the polymerase reads through the integrated HIV
sequence (see reference 1). The HIV sequence is transcribed into the primary
transcript but is degraded along with the rest of the intron after splicing. In
addition, the downstream HIV promoter (in the LTR) may be suppressed by the
active upstream promoter. Perhaps the polymerase reading through the HIV
promoter may disrupt the latter’s ability to bind the correct transcription
factors. This would be overcome when the levels of these factors increases in
activated T4 cells.
As noted above, the transcription factors needed for HIV transcription, such as
NFkB (nuclear factor kappa B) and NFAT (nuclear factor of activated T cells),
may be lacking or restricted to the cytoplasm in resting cells. Many of the
factors that bind to the HIV LTR and increase HIV gene transcription are also
required for transcription of specific genes in an uninfected activated T4 cell,
including NFkB and NFAT. Normally, these factors are cytoplasmic because they
are bound to cytoplasmic retention proteins but they dissociate and the
transcription factors enter the nucleus when the T4 cell is activated. The lack
of NFkB and NFAT in the nucleus does not completely inhibit HIV transcription
but in the absence of TAT, the transcripts are prematurely terminated.
TAT is an HIV-encoded transcription factor but, unlike cellular transcription
factors, it can bind to the DNA promoter of the integrated provirus and to the
RNA transcribed from it. The latter may be the most important. In contrast to
cellular DNA-binding transcription factors, TAT acts at the level of elongation
of the RNA rather than RNA initiation. It binds to a specific secondary
structure in the RNA call the TAT-Responsive element (TAR). After TAT binds to
the TAR, a specific kinase associates with TAT (TAT-associated kinase or TAK)
which consists of two cellular proteins. One of these proteins (called CDK9)
phosphorylates two important targets in the HIV-infected cell. One is RNA
polymerase II, allowing the polymerase to continue elongation of the RNA. The
other is a negative regulatory protein that, on phosphorylation, dissociates
from the TAR. TAT also associates with a variety of other proteins that enhance
HIV RNA transcription.
In the absence of TAT, short transcripts (about 100 nucleotides) are initiated
in the HIV-infected cells but the polymerase usually does not proceed past the
TAR. These short transcripts are found in infected patients who are being
treated with anti-HIV drug cocktails. Some full length HIV RNA molecules are
found but TAT and REV probably do not increase enough to promote high levels of
unspliced HIV RNA in the cytoplasm.
CLINICAL LATENCY
Cellular latency
is different from clinical latency
which refers to fact that symptoms of HIV infection do not manifest themselves as AIDS for
many years (figure 23).
Reference 1:
Lassen K, Han Y, Zhou Y, Siliciano J, Siliciano RF. The multifactorial
nature of HIV-1 latency.
Trends Mol Med. 2004, 10: 525-31.

|

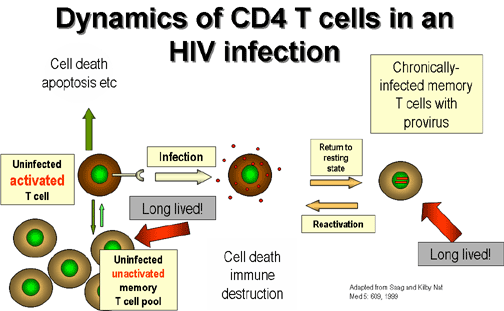 Figure 21
Figure 21